
Erfolgsmodell mit kleinen Schönheitsfehlern
Seit zehn Jahren wird bei allen neuen Medikamenten in Deutschland geprüft, ob sie PatientInnen tatsächlich besser helfen als die bislang verwendeten. Zeit für eine Bilanz.
Am 1. Januar 2011 trat das Arzneimittelmarktneuordnungsgesetz – kurz AMNOG – in Kraft. Hinter dem Wortungetüm steht ein klarer Auftrag: die systematische Prüfung des Nutzens aller Neueinführungen auf dem deutschen Arzneimittelmarkt. Das Gesetz verfolgt zwei Ziele: die Versorgung zu verbessern und die Krankenversicherung finanzierbar zu halten.
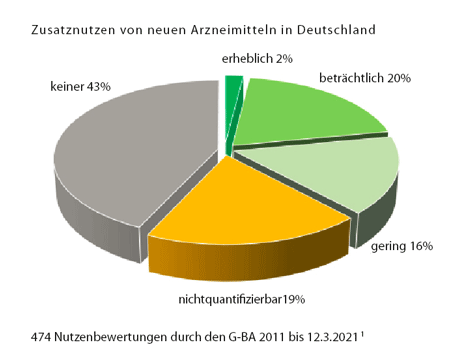
Die wichtigste Erkenntnis aus beinahe 500 Bewertungen (siehe Grafik auf S. 3):[1] Fast die Hälfte der neuen Medikamente (43%), die in den letzten zehn Jahren neu auf den Markt gekommen sind, bieten den PatientInnen keine Vorteile im Vergleich zu der bisher üblichen Behandlung. Gerade einmal 2% sind tatsächlich therapeutische Durchbrüche, immerhin 20% bringen beträchtliche Vorteile, aber 16% sind nur geringfügig besser. Problematisch ist der hohe Anteil der Medikamente mit unklarem Zusatznutzen (19% „nicht quantifizierbar“): Hier ist die Datenlage dermaßen unklar, dass man noch gerade erkennen kann, dass es vermutlich Vorteile gibt, aber sie unmöglich quantifizierbar sind.
All dies bietet jedoch ein zu rosiges Bild. Die genannten Zahlen beziehen sich auf die beste Bewertung eines Wirkstoffs. Häufig bieten neue Medikamente aber nicht allen Erkrankten Vorteile. Wenn die Urteile auf die unterschiedlichen PatientInnengruppen heruntergebrochen werden, findet sich bei rund 60% kein Zusatznutzen.
Dazu kommt: Medikamente gegen seltene Krankheiten (Orphan Drugs)[2] bekommen ohne Prüfung einen Zusatznutzen bescheinigt, solange sie die Krankenversicherung nicht mehr als 50 Millionen Euro pro Jahr kosten. Hier kann der Zusatznutzen also völlig fiktiv sein. Er ist gesetzlich vorgeschrieben, statt auf einer wissenschaftlichen Bewertung zu beruhen. Diese juristisch bedingte Bevorzugung betrifft jede fünfte Bewertung.[3]
Schlechte Studien
Vor allem bei den Waisenmedikamenten ist die Studienlage oft unbefriedigend. Die Zahl der untersuchten PatientInnen ist nicht selten unnötig klein oder die Dauer der Studien zu kurz, was sichere Aussagen über den Nutzen unmöglich macht. Es gibt oft keine Vergleiche gegen andere Behandlungsstrategien, obwohl sie vorhanden sind. Und mitunter wird gar nicht gemessen, was für die Betroffenen tatsächlich spürbare Verbesserungen bringt. Aufgrund dieser Unsicherheiten machen Waisenmedikamente den Löwenanteil der Medikamente mit nicht quantifizierbarem Zusatznutzen aus.
Allerdings wird auch bei anderen neuen Wirkstoffen allzu oft kein direkter Vergleich zu existierenden Medikamenten gemacht. Dadurch ist es fast unmöglich zu beurteilen, ob die Neuerung tatsächlich einen Vorteil bietet. Während sich die Hersteller zu Anfang des AMNOGs noch darauf berufen konnten, dass die Nutzenbewertung viel zu neu sei und die Zulassungsbehörden sich doch mit dem Vergleich gegen Placebo zufriedengegeben hätten, zieht dieses Argument heute nicht mehr – es wird von den Firmen aber trotzdem immer noch bemüht. Ein Trend zu mehr direkt vergleichenden Studien ist leider nicht zu erkennen. Insofern vergoss der Chef des Pharmaverbandes Vfa kürzlich Krokodilstränen, als er beklagte, dass fast die Hälfte der Studien vom G-BA für die Nutzenbewertung nicht berücksichtigt würde.[4] Das liegt nämlich einfach daran, dass sie dafür schlicht ungeeignet sind. Über die Jahre liegen für rund die Hälfte der neuen Medikamente keine direkten Vergleiche zum bisherigen Therapiestandard vor.[5]
Ein Problem sind auch die oft verfrühten Zulassungen bei noch gar nicht abgeschlossenen Studien. Solche Zwischenauswertungen lassen die Neuerungen manchmal in einem zu positivem Licht erscheinen. Zwei krasse Fälle betrafen zwei Krebsmedikamente, die vom G-BA einen Zusatznutzen zugesprochen bekamen. Später stellte sich heraus, dass der Wirkstoff Olaratumab unwirksam ist[6] und Ingenolmebutat Hautkrebs sogar häufiger macht.[7] Beide Mittel wurden verboten.
Her mit den Daten
Deutschland war mit der Einführung einer Nutzenbewertung spät dran. Andere europäische Staaten haben einen solchen Realitäts-Check schon seit vielen Jahren. Dafür bietet das deutsche Gesetz aber einen bedeutsamen Vorteil im Vergleich zu unseren Nachbarländern. Es verpflichtet nämlich den Anbieter, die vollständigen Studiendaten zu seinem Produkt zur Bewertung einzureichen, also auch den Teil, der gewöhnlich selbst für die Fachwelt unsichtbar bleibt.
Um sich ein Bild von solchen Geheimniskrämereien zu machen, ein Vergleich: Aus allen öffentlichen Quellen zusammengenommen[8] sind gerade einmal gut die Hälfte aller Studienergebnisse verfügbar, im AMNOG-Verfahren sind es dagegen nahezu 90%.[9] Das erlaubt eine viel fundiertere Einschätzung zum tatsächlichen Nutzen eines neuen Präparats.
Transparenz macht den Unterschied
Dazu ein Beispiel: Während das für England und Wales zuständige NICE dem Brustkrebsmedikament Palbociclib einen Zusatznutzen zusprach, konnte der G-BA keine Vorteile entdecken. Der Unterschied: Bei den Nachbarn lagen nur unvollständige Informationen zu dem Medikament vor. Der Hersteller behauptete, er hätte noch keine Daten dazu, ob die Frauen durch das neue Medikament tatsächlich länger überleben, aber bestimmte Messwerte gäben einen starken Hinweis dafür. Aufgrund der vollständigen Daten konnte in Deutschland demgegenüber errechnet werden, dass kein Überlebensvorteil durch Palbociclib vorhanden war. In England wurden die entscheidenden Angaben für die Bewertung vor der Veröffentlichung der Daten geschwärzt.[10] Es bleibt deshalb unklar, welche Zahlen dem NICE für die Entscheidung vorlagen. Die Begründung der Behörde bezog sich nicht auf einen Überlebensvorteil, sondern lediglich auf die besseren Messwerte, offensichtlich ein Trugschluss.
Verbesserungen angestoßen
In einem Punkt haben die Anbieter durch das Nutzenbewertungsverfahren auf jeden Fall dazugelernt. Während es zu Beginn des AMNOG-Verfahrens nur selten Daten zur Lebensqualität gab, legen heute drei Viertel der Anbieter auch Angaben vor, ob sich die PatientInnen durch die Behandlung mit dem neuen Präparat auch wirklich besser fühlen. Allerdings geschieht das oft noch halbherzig: Denn wenn die Lebensqualität nur bis zum Zeitpunkt einer Verschlechterung der Erkrankung gemessen wird (also z.B. zum Zeitpunkt, wo sich bei Krebs Metastasen bilden), fehlen wesentliche Informationen. Für die Erkrankten ist es aber sehr wichtig zu wissen, wie gut es ihnen in der verbleibenden Lebensspanne noch gehen mag.
Kräftig gespart
Finanziell hat die Nutzenbewertung die Krankenkassen und damit alle Versicherten deutlich entlastet. Zwar wurde erst nach acht Jahren, 2018, die vom Gesetzgeber angepeilte jährliche Ersparnis von zwei Milliarden Euro erreicht, für 2020 wurden aber durch die ausgehandelten Rabatte schon fast vier Milliarden Euro eingespart. Trotzdem bleiben die rasant steigenden Preise für neue Arzneimittel, die inzwischen bis in den Millionenbereich pro PatientIn reichen, ein Problem.
Was kommt in der Praxis an?
Ein Kritikpunkt am AMNOG-Verfahren ist, dass die detaillierten Erkenntnisse darüber, welche Medikamente für welche Zielgruppen Vorteile bringen, in der Krankenversorgung nicht ankommen. Ein Indiz dafür ist, dass auch neue Medikamente, die keinerlei Zusatznutzen haben, häufig verordnet werden. Damit sich das ändert, hat der Gesetzgeber verfügt, dass die Ergebnisse der Nutzenbewertung auf den Computern in Arztpraxen dargestellt werden müssen. Auch PatientInnen können sich selber schlau machen. Auf den Seiten des IQWiG gibt es verständliche Zusammenfassungen der Vor- und Nachteile von neuen Arzneimitteln.[11]
 Verbesserungen möglich
Verbesserungen möglich
Um die Preisspirale zu durchbrechen, gibt es mehrere Vorschläge. Der wichtigste ist vielleicht die rückwirkende Preisfestsetzung. Denn bislang beginnt der ausgehandelte Rabatt für ein neues Arzneimittel erst nach zwölf Monaten. Auf diese Weise können Anbieter – auch die, die ein Mittel ohne jeden Zusatznutzen auf den Markt bringen – ein Jahr lang kräftig absahnen.
Ein besonderes Problem sind die teuren Krebsmedikamente. Wenn die Vergleichstherapie schon 50.000 € oder mehr kostet, bedeutet ein neues Mittel für den Anbieter selbst dann ein gutes Geschäft, wenn wegen fehlenden Zusatznutzens kein höherer Preis erzielt werden kann.
Ein weiterer Vorschlag betrifft den oft enorm hohen Anfangspreis. Firmen sollen offenlegen, wie viel Geld sie in die Forschung gesteckt haben – und inwieweit ihr Produkt auch auf den Ergebnissen staatlich geförderter Forschung an Universitäten und öffentlichen Instituten beruht.
Auch die Nutzenbewertung sollte in einigen Punkten strenger werden. Der Freibrief für Waisenmedikamente unter 50 Millionen Euro Jahresumsatz muss entfallen, und Arzneimittel, die unter Auflagen zugelassen wurden, sollten immer nur eine befristete Bewertung erhalten.[12] Bei solchen Arzneimitteln kann der G-BA inzwischen vom Anbieter weitere anwendungsbegleitende Studien verlangen, um die Erkenntnislage zu verbessern. Bislang fehlt es aber an Sanktionsmöglichkeiten, wenn diese Auflage nicht erfüllt wird.
Undurchsichtige Expertise
Ein weiterer Schwachpunkt des Verfahrens bleibt voraussichtlich noch länger erhalten: Die Intransparenz bei den an Anhörungen beteiligten ExpertInnen. Sie stellen einen wichtigen Beeinflussungsfaktor bei der Nutzenbewertung dar. Während bei den IndustrievertreterInnen offensichtlich ist, welche Interessen sie vertreten, bleibt bei VertreterInnen von Fachgesellschaften die Verstrickung mit den Anbietern der diskutierten Arzneimitteln undurchsichtig – und nicht wenige pflegen enge Kontakte zu den Firmen. Es müssen bei den Anhörungen zwar detaillierte Erklärungen zu Interessenkonflikten vorgelegt werden, diese sind aber nur in absolut kondensierter Form auf der Website des G-BA einzusehen. So kann die Öffentlichkeit nicht mehr erkennen, dass Professor X zwar offiziell eine Fachgesellschaft vertritt, aber gleichzeitig dem Hersteller als Berater für genau das Produkt dient, über dessen Nutzen gerade diskutiert wird. Als Argument gegen eine vollständige Veröffentlichung wird der Schutz persönlicher Daten vorgebracht. Aber oft hat derselbe Professor gar kein Problem, (einen Teil seiner) Interessenkonflikte in internationalen Fachzeitschriften relativ detailliert anzugeben, weil diese sonst den Artikel schlicht nicht veröffentlichen würden.
Bei allen Fortschritten, die das AMNOG gebracht hat, gibt es also noch einigen Verbesserungsbedarf. (JS)
Eine kürzere Vorfassung dieses Artikels erschien in Gute Pillen – Schlechte Pillen (2021) Nr. 3, S. 18
Screenshot Nutzenbewertung ©Auszug aus dem Pharma-Brief
Grafik Zusatznutzen © www.g-ba.de/service/veranstaltungen/10-jahre-amnog
Artikel aus dem Pharma-Brief 3-4/2021, S.1
[1] G-BA-Tagung „10 Jahre AMNOG“ am 19.3.2021 www.g-ba.de/service/veranstaltungen/10-jahre-amnog
[2] Orphan Drugs (Waisenmedikamente) Arzneimittel gegen Krankheiten, von denen nicht mehr als 5 von 10.000 Menschen betroffen sind
[3] Bewertungen bis 31.12.2019. Storm A (Hrsg.) (2020) AMNOG-Report 2020. S. 215
[4] Han Steutel (Vfa) auf der G-BA-Tagung „10 Jahre AMNOG“ am 19.3.2021
[5] Thomas Kaiser (IQWiG) auf der G-BA-Tagung „10 Jahre AMNOG“ am 19.3.2021
[6] Pharma-Brief (2019) Zu früh ist unzuverlässig. Nr.1, S. 4
[7] Pharma-Brief (2020) Ungesunde Eile. Nr. 2, S. 6
[8] Veröffentlichung in Fachzeitschriften, Bericht der Zulassungsbehörde, Studienregister
[9] Untersucht anhand von 15 AMNOG-Verfahren. Köhler M. u.a. (2015) BMJ; 350, S. h796
[10] Pharma-Brief (2019) Die nächste Schlappe. Nr. 2, S. 6
[11] www.gesundheitsinformation.de/vom-iqwig-bewertete-wirkstoffe.html
[12] Für Orphan Drugs mit bis zu 50 Mio. € Umsatz führt der G-BA selbst eine Teilbewertung durch.