
2019-artikel
Zu früh ist unzuverlässig
Warum vorschnelle Zulassungen ein Problem sind
Neue Arzneimittel kommen immer öfter auf dünner wissenschaftlicher Basis auf den Markt. Der Fall des Krebsmedikaments Olaratumab macht schlaglichtartig die Probleme deutlich. Nach Abschluss einer zweiten Studie verfügte die europäische Zulassungsbehörde EMA, dass keine weiteren PatientInnen mit dem Medikament behandelt werden dürfen.[1]
Olaratumab wurde im November 2016 zugelassen und sollte gegen seltene bösartige Tumore helfen, sogenannte Weichteilsarkome. Die Pharmazeutische Zeitung bescheinigte der EMA ein „rekordverdächtiges Tempo“ und war voll des Lobes. Es wurde ein Experte zitiert, der die laut Studie um ein Jahr verlängerte Lebensdauer als „absolut sensationell“ bezeichnete.[2] Der Haken an der Sache: statt einer Phase III Studie, die üblicherweise für die Zulassung verlangt wird, hatte der Hersteller nur eine kleine Phase Ib/II Studie mit etwas über 100 Teilnehmern durchgeführt. Diese frühen Studien dienen eigentlich nur der Prüfung der Verträglichkeit und der Dosisfindung.
Ganz sicher war sich deshalb die europäische Arzneimittelbehörde EMA auch nicht und ließ Olaratumab nur unter der Bedingung zu, dass der Hersteller eine weitere größere Studie durchführt. Erste Ergebnisse der Phase III-Studie mit über 400 ProbandInnen sickerten jetzt durch: Es gibt keinerlei Überlebensvorteil durch das neue Medikament. Und auch die Vorteile bei der oft als Hilfskonstruktion eingeführten Messgröße „progressionsfreies Überleben“ – ein fragwürdiges Surrogat für patientenrelevante Vorteile – brachen in sich zusammen. In der Phase III-Studie fand sich kein Unterschied zur Vergleichstherapie.
Die Behörde verfügte, dass Olaratumab bis zur endgültigen Auswertung der Studie keinen neuen PatientInnen verordnet werden darf.[1] Die Jahrestherapiekosten betragen in Deutschland 186.448,86 €. Das Präparat muss zusammen mit der bislang üblichen Therapie Doxorubicin (Preis 3.200,56 € jährlich) gegeben werden.[3] Laut EMA wurden in Europa bislang tausend Erkrankte behandelt. Für die PatientInnen mehr Nebenwirkungen, für den Hersteller ein gutes Geschäft. Lilly erzielte mit dem Präparat bis Ende 2018 über eine halbe Milliarde US$ Umsatz.[4],[5]
Der Fall erinnert an das Brustkrebsmedikament Palbociclib, das auf Basis von noch laufenden Studien zugelassen worden war. Der Hersteller hatte auf Basis des längeren progressionsfreien Überlebens hohe Erwartungen geschürt. Die versprochene Lebensverlängerung wurde anschließend in zwei Studien nicht bestätigt.[6]
Artikel aus dem Pharma-Brief 1/2019, S.4
[1] EMA (2019) No new patients should start treatment with Lartruvo. Press release 23.1.
[2] Mende (2016) Pharmazeutische Zeitung Nr. 47 www.pharmazeutische-zeitung.de/ausgabe-472016/rekordverdaechtiges-tempo
[3] G-BA (2017) Nutzenbewertung von Olaratumab. Beschluss vom 18.5. www.g-ba.de/informationen/nutzenbewertung/268
[4] Lilly (2018) Q4_2017 workbook
[5] Lilly (2019) IR workbook Q4 2018
[6] Pharma-Brief (2018) Brustkrebs: Leere Versprechen. Nr. 6, S. 4
Wohin das Geld fließt
Schwachstellen bei Arzneimittelversorgung
Längst nicht alles, was in Deutschland zu Lasten der Krankenkassen verordnet wird, ist auch sinnvoll. Und oft steht der Preis in keinem Verhältnis zum Nutzen. Das macht der Arzneiverordnungs-Report 2019 (AVR) deutlich.
41,2 Mrd. € gaben die gesetzlichen Krankenkassen 2018 für Arzneimittel aus,[1] das sind 3,2% mehr als im Vorjahr. Dabei machten patentgeschützte Arzneimittel nur 6,6% aller verordneten Tagesdosen aus, verursachten aber fast die Hälfte der Kosten (46,2%) – Tendenz rasch steigend. Von 2008 bis 2018 verdreifachte sich der Preis pro Rezept. Deshalb setzt sich der AVR auch besonders intensiv mit diesem Marktsegment auseinander.
Eine Analyse der 2018 neu zugelassenen Arzneimittel macht den Sprengstoff deutlich: Würden alle PatientInnen, die für eine Behandlung in Frage kommen, diese neuen Mittel verschrieben bekommen, würde das die Kassen 52,9 Mrd. € kosten[2] – also mehr als bislang insgesamt für Arzneimittel ausgegeben wird.
Dabei hat jedes dritte Medikament laut Bewertung des Gemeinsamen Bundesausschusses (G-BA) keinen Zusatznutzen. Und selbst wenn Vorteile erkennbar sind, profitieren häufig nur bestimmte PatientInnengruppen. Das gilt zum Beispiel für Erenumab, das zur Migräneprophylaxe eingesetzt wird. 2,4 Millionen MigränepatientInnen gibt es. Würden alle Erenumab gespritzt bekommen, müssten die Kassen dafür unfassbare 30,3 Mrd. € zahlen. Vorteile bietet das Medikament aber nur für eine sehr kleine Gruppe: rund 14.000 -15.000 Personen, denen andere Mittel zur Vorbeugung nicht geholfen haben.
Wobei die Bewertung neuer Arzneimittel alles andere als einfach ist. Oft winkt die europäische Zulassungsbehörde EMA neue Medikamente auf Basis von spärlichen Daten durch. Da wird nur gegen Placebo getestet, statt gegen die etablierte Therapie. Oder es wird gar nicht gemessen, was den PatientInnen wirklich nützt, sondern nur die Verbesserung von Laborwerten oder anderen Surrogaten. Besonders deutlich wird die Evidenzlücke im Bereich der Krebsmedikamente, die oft als Orphan Drugs – also Medikamente gegen seltene Erkrankungen – auf den Markt kommen. Warum in diesem Bereich auf EU-Ebene gesetzliche Verbesserungen notwendig sind, das analysiert der AVR ausführlich.
Es ist aber nicht alles schwarz: Durch Arzneimittelfestbeträge konnten 2018 Einsparungen in Höhe von 8,2 Mrd. € erzielt werden und auch die Preisverhandlungen nach den Nutzenbewertungen entlasteten die Versicherten um 2,7 Mrd. €.
Artikel aus dem Pharma-Brief 10/2019, S. 7
Titelbild Arzneiverordnungs-Report 2019 © Springer
[1] Einschließlich Zuzahlungen der Versicherten
[2] Berechnet für die 30 Mittel, für die eine Nutzenbewertung vorliegt.
Wo bleibt der Nutzen?
IQWiG zieht Bilanz nach 7 Jahren Bewertung
Das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) kommt zu einem ernüchternden Urteil: Nur wenige neue Arzneimittel bringen PatientInnen substanzielle Vorteile. Das Institut bleibt aber nicht bei einer Analyse stehen, es fordert eine Umorientierung der Medikamentenforschung.[1]
Seit 2011 müssen alle neuen Medikamente, die in Deutschland auf den Markt kommen, auf ihren Zusatznutzen bewertet werden. Für die wissenschaftliche Bewertung ist mit wenigen Ausnahmen[2] das IQWiG zuständig, die endgültigen Entscheidungen trifft der Gemeinsame Bundesausschuss (G-BA).[3] Von 2011 bis 2017 wurden 216 Medikamente durch das Institut bewertet, davon waren 152 neue Wirkstoffe und 64 neue Indikationen. Für 125 der Medikamente (58%) wurden keine Daten vorgelegt, die eine Verringerung der Sterblichkeit, der Symptome oder Verbesserung der Lebensqualität bestätigt hätten. Zwei Mittel (1%) waren sogar schlechter als die etablierte Vergleichstherapie.
Nur einem Viertel der Neueinführungen (54 Mittel) konnte ein beträchtlicher oder erheblicher Zusatznutzen bescheinigt werden. 16% (35) hatten einen geringen oder nicht quantifizierbaren Zusatznutzen. Ein genauerer Blick relativiert selbst diese Zahlen. Denn bei knapp der Hälfte (42%) dieser Mittel konnte eine Verbesserung nur für bestimmte PatientInnengruppen festgestellt werden.
Am schlechtesten schneiden Psychopharmaka (nur 6% mit Zusatznutzen) und Mittel gegen Diabetes ab (17% mit Zusatznutzen). Auffällig ist das Ungleichgewicht bei den Anwendungsgebieten: über ein Drittel (82) sind Krebsmedikamente. Diese schneiden bei der Bewertung deutlich besser ab, 59% konnte ein Zusatznutzen bescheinigt werden. Ähnliches gilt für die zweitgrößte therapeutische Gruppe, den 29 Mitteln gegen Infektionen (vor allem Hepatitis C und HIV), 62% mit Zusatznutzen.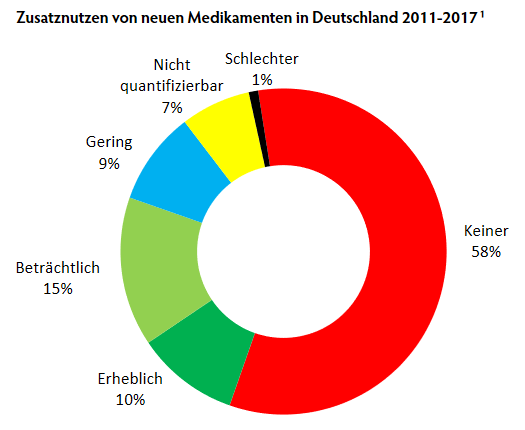
Ungewissheit
Was verbirgt sich hinter der großen Gruppe von Medikamenten, denen kein Zusatznutzen bescheinigt werden konnte? In vielen Fällen sorgen ungeeignete Studien für ein negatives Urteil. Wenn nur gegen Placebo getestet wurde (64 Mittel), statt gegen eine Vergleichstherapie, kann man gar nicht beurteilen, ob das Medikament schlechter, besser oder gleichgut wirkt. Ebenfalls häufig (42 Mittel) war in den Studien ein ungeeignetes Vergleichsmedikament gewählt worden (für die Indikation gar nicht zugelassen, ungeeignete Dosis gewählt u.a.). 19 Medikamente waren nicht besser als die etablierte Therapie.
Auch die Hoffnung, dass nach Zulassung weiter geforscht wird und sich die die Ungewissheit dadurch auflöst, erfüllt sich selten. Entweder werden geeignete Studien gar nicht erst durchgeführt oder die Endergebnisse[4] bzw. besser gemachte Studien zeigen dann keinen zusätzlichen Nutzen.
Auf jeden Fall macht die Ungewissheit über den Nutzen bei Zulassung vernünftige Therapieentscheidungen schwierig.
Mehr vom Gleichen
Auffällig ist die große Ähnlichkeit etlicher erfolgreicher neuer Medikamente, die auf dem gleichen Wirkprinzip basieren. So sind ein Viertel aller positiv bewerteten Krebsmedikamente PD-1 oder PD-L1 Inhibitoren, weitere befinden sich in der Forschungspipeline. Alle neuen Hepatitis C-Medikamente basieren auf direkter antiviraler Wirkung. Das stellt eine Verschwendung von Forschungsgeldern dar (die die Allgemeinheit über hohe Medikamentenpreise refinanziert).
Neue Lösungen gefragt
Die drei MitarbeiterInnen des IQWiG formulieren klare Änderungsvorschläge. Als erster Schritt wird ein Umdenken bei der Zulassung gefordert. Es sollten wieder ausreichend große Phase III-Studien durchgeführt werden, die Ergebnisse messen, die für PatientInnen relevant sind (und nicht nur die Verbesserung von Laborwerten). Um Daten für eine Nutzenbewertung zu gewinnen, sollte – wo möglich – gegen die etablierte Therapie verglichen werden. Bei Entscheidungen über die Erstattung und Preise dürften marginale Fortschritte und unsichere Ergebnisse nicht belohnt werden.
Langfristig müsse die Gesundheitspolitik eine aktivere Rolle übernehmen: Statt passiv darauf zu warten, was die Pharmaindustrie entwickelt, sollte die Gesellschaft selbst Prioritäten für die Forschung setzen. Ein Vergleich mit der Entwicklungspipeline der Industrie mit dem tatsächlichen Bedarf kann Lücken identifizieren. Öffentlich finanzierte Forschungsprogramme können eine wirksame Maßnahme sein. Das beweist für die vernachlässigten Krankheiten bereits seit einiger Zeit die Drugs for Neglected Diseases initiative (DNDi), die erfolgreich Medikamente für Tropenkrankheiten entwickelt. Im Bereich der Antibiotikaforschung sind die WHO und auch die Bundesregierung aktiv geworden. Auch andere Modelle wie Forschung als öffentliches Unternehmen oder Open Source böten vielversprechende Möglichkeiten. (JS)
Artikel aus dem Pharma-Brief 4-5/2019, S.13
[1] Wieseler B et al. (2019) New drugs: where did we go wrong and what can we do better? BMJ; 366, p i4340
[2] Medikamente gegen seltene Krankheiten (Orphan Drugs) werden bis zu einem Jahresumsatz von 50 Mio. € durch den G-BA selbst bewertet. Dabei gilt aufgrund der gesetzlichen Vorgaben (AMNOG) ein fiktiver Zusatznutzen als belegt. Der G-BA kann nur noch das Ausmaß festlegen bzw. entscheiden, dass er einen Zusatznutzen nur aus rein rechtlichen Verpflichtungen erteilt.
[3] Pharma-Brief (2013) Ein Erfolgsmodell soll abgeschafft werden. Nr. 8-9, S. 6
[4] Pharma-Brief (2019) Die nächste Schlappe. Nr. 2, S. 6
Wir kennen die Realität nicht
Wenig Daten zu Antibiotika-Resistenzen weltweit
Im Oktober 2015 führte die WHO ein standardisiertes System zur internationalen Überwachung antimikrobieller Resistenzen ein. Das Global Antimicrobial Resistance Surveillance System (GLASS) soll verlässliche Daten zur weltweiten Resistenzproblematik liefern und damit nationale, regionale und globale Aktionspläne unterstützen. Aber wie gut funktioniert die Datenerhebung und wie verlässlich sind die gewonnenen Informationen? Welche Probleme gibt es bei der Implementierung von GLASS in einem armen Land wie Nigeria. Und wo gibt es noch Baustellen in Deutschland? Dr. Gerhard Schwarzkopf Steinhauser – Experte im Bereich Mikrobiologie und Infektionsschutz stand uns dazu Rede und Antwort.
Seit 2016 können Daten zu antimikrobiellen Resistenzen in die GLASS Datenbank eingespeist werden. 72 Länder nehmen inzwischen an dem Programm teil. Wissen wir nun mehr über die globale Resistenzlage?
Etliche Länder haben GLASS zwar ratifiziert und sich damit verpflichtet, nach einem vorgegebenen Schema Daten zu liefern. Die Datengrundlage ist aber ziemlich lückenhaft, entspricht nicht den Qualitätsanforderungen und ist in keinster Weise repräsentativ.
Wo hakt es bei der Datenerhebung?
Entscheidend ist, dass die Resistenztestung nach bestimmten Qualitätsstandards erfolgt – GLASS empfiehlt dazu die europäische Methode EUCAST oder das amerikanische CLSI System. Die Resistenztestung muss somit auf streng definierten Medien und mit ganz bestimmten labortechnischen Methoden erfolgen. Wissen und Technik sind aber in vielen Krankenhäusern nicht vorhanden – das gilt ganz besonders für arme Länder. Nigeria z.B. hat GLASS ratifiziert und ist dabei, in Kooperation mit dem RKI die Datenerhebungen zu beginnen. Dafür müssen aber zunächst die Voraussetzungen geschaffen werden. Selbst wenn die nötige Technik verfügbar ist, gibt es derzeit noch erhebliche Lücken beim Wissensstand des Personals in den mikrobiologischen Labors.
Sie halten die erhobenen Daten außerdem für wenig repräsentativ …
In Nigeria müssen Patienten die Diagnostik selbst bezahlen, deshalb wird nicht in ausreichender Menge diagnostiziert. Wenn z.B. bei jedem komplizierten Harnwegsinfekt eine mikrobiologische Testung gemacht werden würde, dann wäre die Datenlage repräsentativ. Wenn die Testung vom Geldbeutel der Patienten abhängt, gilt das nicht.
Ein Problem ist auch in vielen Ländern die Trennung des Gesundheitssystems in einen öffentlichen und einen privaten Sektor. Die GLASS Datenbank unterscheidet nicht zwischen den Daten aus öffentlichen und privaten Gesundheitseinrichtungen. Sie gibt auch nicht vor, welche oder wie viele Einrichtungen Daten liefern sollen. Festgeschrieben ist nur, dass die Auswahl repräsentativ sein soll. Das ist ein Manko. Wenn man Daten erhebt, muss man wissen, wo die herkommen – schließlich ist bei armen Bevölkerungsteilen eine andere Epidemiologie zu erwarten als bei reichen Bevölkerungsgruppen. Schon aufgrund der Lebensweise, des Zugangs zu Trinkwasser und der Ernährungsgewohnheiten sind möglicherweise die Erreger bei bakteriellen Infektionen höchst unterschiedlich.
Und wie steht es nun in Deutschland und Europa?
Es gibt Länder, wo die Datenerfassung streng geregelt ist – etwa in den Niederlanden oder in den skandinavischen Ländern inklusive Dänemark. In Deutschland gibt es dagegen gewaltige Schwächen. Zwar ist die Erhebung von Daten zum Antibiotika-Verbrauch und zur Resistenzlage im Infektionsschutzgesetz gesetzlich vorgeschrieben. Aber die Übermittlung dieser Daten an die nationale Datenbank zur Antibiotika-Resistenz-Surveillance (ARS) des RKI ist freiwillig. Die ARS-Daten sind daher derzeit nicht repräsentativ. Eine Verpflichtung zur Datenübermittlung ist wiederum Ländersache. Bayern will beispielsweise eine eigene Datenbank einrichten, was nicht zielführend ist.
Was wäre die Lösung für all diese Probleme?
Zweierlei: Eine vernünftige Diagnostik und eine Verpflichtung zur Übermittlung sämtlicher Daten. Für arme Länder heißt das auch, einen nationalen Gesundheitsservice zu etablieren, der eine gute Versorgung für alle garantiert. Denn nur ein guter Zugang zu Gesundheitsdienstleistungen und zu Diagnostika ermöglicht eine gute Kenntnis des Gesundheitszustands und der Resistenzlage innerhalb der Bevölkerung. Bisher wissen wir nicht, wie gravierend die Resistenzraten tatsächlich sind. Dazu müssten wir die Realität besser kennen.
Artikel aus dem Pharma-Brief 4-5/2019, S.14
Bild Dr. Schwarzkopf Steinhauser © Jörg Schaaber
[1] Global Health Protection Programme des Bundesministeriums für Gesundheit: NiCaDe https://ghpp.de/de/projekte/nicade [Zugriff 24.7.2019]
[2] Nigeria Centre for Disease Control (2019) NCDC and RKI Launch New Project to Strengthen Health Security in Nigeria, 13 June https://ncdc.gov.ng/news/181/ncdc-and-rki-launch-new-project-to-strengthen-health-security-in-nigeria [Zugriff 23.7.19]
Wie man es besser machen kann
Die Universität Nottingham sorgt für Transparenz bei Studien
Die Universität Nottingham ist ein europäischer Vorreiter in Sachen Transparenz bei klinischen Studien. Von über 95% der fälligen Studien wurde mittlerweile eine Zusammenfassung der Ergebnisse im europäischen Studienregister EudraCT eingetragen – ein steiler Anstieg verglichen mit 2018, damals waren nur 8% veröffentlicht. Till Bruckner von TranspariMED fragte die Universität, wie sie mit ihren ungemeldeten Studien umgegangen ist und was andere europäische Universitäten davon lernen können.
Wann habt ihr angefangen, systematisch die fehlenden Studienergebnisse in EudraCT hochzuladen?
Die Universität Nottingham hat ihre Studienregistrierungen in EudraCT im Dezember 2018 geprüft und unmittelbar angefangen fehlende Ergebnisse hochzuladen oder zu aktualisieren. Dieser Prozess war im März 2019 abgeschlossen.
Zur gleichen Zeit hat die Universität ihre Richtlinien, Systeme und Prozesse überprüft und überarbeitet, um sicherzugehen, dass die zusammengefassten Ergebnisse von den verantwortlichen WissenschaftlerInnen künftig rechtzeitig den entsprechenden Registern gemeldet werden. Ein neuer Leitfaden zur Studienregistrierung wurde am 17. Dezember 2018 veröffentlicht.
Dieser Leitfaden wurde regelmäßig dem wissenschaftlichem, forschendem Personal kommuniziert. Gleichzeitig gab es Unterstützung von unserem Research Governance Team, um bei der angemessenen Studienregistrierung und Ergebnismeldung zu helfen.
Wie viele Ergebnisse haben gefehlt, als ihr angefangen habt und wie viele habt ihr seitdem hochgeladen?
46 Studienergebnisse wurden während der Überprüfung als potenziell fehlend ermittelt. Bei einer stellte sich heraus, dass es sich um keine klinische Studie handelte, eine weitere Studie wurde zwar registriert, aber gar nicht durchgeführt. Diese Angaben wurden korrigiert und alle anderen Studienergebnisse wurden hochgeladen.
Wie ist der Prozess organisiert? Wer macht was?
Die Leiterin der Research Governance hat detailliert jeden Registereintrag geprüft und eine ausführliche Analyse zu den nicht gemeldeten Ergebnissen durchgeführt und den verantwortlichen Studienleiter identifiziert. Alle wurden von der Leiterin der Research Governance und dem Vize-Kanzler der Fakultät mit der Bitte kontaktiert, die Ergebnisse zu veröffentlichen. Gleichzeitig wurde Unterstützung und Beratung angeboten.
Um den Rückstand schnellstmöglich aufzuholen hat die Leiterin der Research Governance in einer beachtlichen Zahl von Fällen die Ergebnisse von Studienleiter direkt erhalten. Anschließend hat sie bei der europäischen Zulassungsbehörde EMA um den Transfer der Studieneinträge zu ihrem Benutzerkonto gebeten. Anschließend hat sie das Register aktualisiert und dann die britische Zulassungsbehörde MHRA[1] kontaktiert, damit dieses den Status der Studien in EudraCT zu „abgeschlossen“ ändert.
Wie seid ihr mit alten Studien umgegangen, die fälschlicherweise als „laufend“ im EudraCT gelistet waren?
Wenn Studien fehlerhaft registriert oder fälschlich als „laufend“ gelistet waren, hat die Leiterin der Research Governance sich in Verbindung mit der EMA und dem MRHA gesetzt, um den Eintrag zu korrigieren.
Welche Ressourcen wurden benötigt? Musste zusätzliches Personal angestellt werden? Wie lange dauerte es pro Studie?
Bis heute wurde die Überprüfung und die nachfolgenden Prozesse überwiegend durch die Leiterin der Research Governance überwacht und durchgeführt.Zurzeit bekommt das Research Governance Team zwei zusätzliche Stellen, um die Qualität im Datenmanagement sowie bei der Überprüfung der Studien im Forschungsportfolio der Universität weiter zu fördern. Außerdem sollen sie die Forschenden unterstützen, ihrer Verantwortung bei der Studienregistrierung und Ergebnismeldung nachzukommen.
Die benötigte Zeit um jeden EudraCT Eintrag zu aktualisieren hing sehr von der Verfügbarkeit und dem Format der Daten ab, sowie von den Herausforderungen beim Hochladen im Register und der nachfolgenden Zusammenarbeit mit der EMA und dem MHRA. Im Durchschnitt brauchte es für jeden Eintrag mehrere Stunden.
Was waren die Haupthindernisse, auf die ihr gestoßen seid und wie habt ihr sie bewältigt?
Häufig mussten die wissenschaftlichen Daten und Tabellen neu formatiert werden, um mit EudraCT übereinzustimmen. Die WissenschaftlerInnen oder die Leiterin der Research Governance mussten die Daten per Hand in das geeignete Format umwandeln, das war sehr zeitraubend.
Da viele WissenschaftlerInnen und Studierende an den klinischen Studien beteiligt waren und persönlich für die Studienregistrierung und Ergebnisübermittlung verantwortlich waren, war die Überwachung und Aktualisierung der zentralen Einträge durch Research Governance Team nicht einfach. Um diese Schwierigkeit zu umgehen, hat die Leiterin ein Standardvorgehen eingeführt. Sie erhält von den Forschenden die EudraCT Nummer und die Registrierung für CTIMPs,[2] damit alle künftigen Studien unter ihrem Account registriert werden. Dadurch wird verhindert, dass ungewollt bei einem Account Details verloren gehen. Gleichzeitig wird die zentrale Überwachung unterstützt und es wird sichergestellt, dass die WissenschaftlerInnen, die die klinischen Studien durchführen, daran erinnert werden, Ergebnisse zu melden und zu aktualisieren.
Das Research Governance Team pflegt eine Datenbank mit allen Studien, die gemäß den gesetzlichen Vorgaben und Vorschriften des Gesundheitsministeriums als klinische Studien gemeldet werden müssen. Allerdings führt die Universität auch viele andere Forschungsvorhaben durch, für die diese Kriterien nicht gelten. Das Governance Team hat keinen Überblick über solche Studien. Momentan überprüfen wir, wie wir auch diese Studien zukünftig verfolgen können.
Was sind die drei wichtigsten Dinge, die andere Verantwortliche tun können, um die Studienmeldung für Universitäten zu vereinfachen?
EudraCT, wie auch die meisten anderen Register, bietet keine Möglichkeit, den größeren Kontext darzustellen, in dem eine Studie stattfindet, Ergebnisse zu diskutieren sowie nächste Schritte zu besprechen. Die Einführung eines gemeinsamen Formats innerhalb oder besser noch registerübergreifend würde die oben genannten Formatierungsprobleme lösen. Weitere Funktionen, um Inhalte sowie nächste Schritte festzuhalten, könnten den Nutzen für und die Forschungsgemeinschaft erhöhen und die Compliance verbessern.
Es gibt keine eindeutige Definition, was genau zu klinischer Forschung zählt – trotz des Versuchs der WHO eine Definition zu finden.[3] Um den Anforderungen des International Committee of Medical Journal Editors zu genügen,[4] werden Register auch für nicht-klinische (aber medizinische) und physiologische Studien benutzt.
Das ist die Voraussetzung dafür, dass die Ergebnisse in den Fachzeitschriften auch veröffentlicht werden können, unabhängig davon, ob die Studie tatsächlich als klinische Forschung definiert wurde oder nicht. Es gibt Unklarheiten, was tatsächlich in einem öffentlichen Register eingetragen werden muss und was nicht.
Viele dieser Studien sind Studierendenprojekte, die oftmals keine neuartige Forschung beinhalten und wenig zu der medizinischen Wissensbasis beitragen. Deshalb würden die Register gut daran tun, einen Mechanismus zu haben, mit dem die Forschenden und Sponsoren angeben können, um welchen Studientyp es sich handelt und ob beabsichtigt ist, die Ergebnisse zu veröffentlichen oder die Wissensbasis zur klinischen Anwendung zu erweitern.
Bisher haben Register außerdem keinen Mechanismus, der Einträge löscht oder den Studienstatus verändert, wenn eine Studie nie begonnen oder so frühzeitig gestoppt wurde, dass es keine Ergebnisse zu melden gibt.
Basierend auf eurer Erfahrung, was würdet ihr anders machen, wenn ihr den gesamten Prozess heute noch einmal starten würdet?
Wir würden die Maßnahmen ergreifen, die wir erst im Laufe des Prozesses erarbeitet haben, insbesondere die erweiterte zentrale Prüfung, Analyse- und Überwachungsprozeduren.
Außerdem würden wir die Forschungsgemeinschaft besser in die Bedeutung der Pflege der Registereinträge einbeziehen. Die Leiterin der Research Governance wird nun regelmäßige Überprüfungen durchführen, auf Fakultätrats- und Komiteeebene aktuelle Informationen liefern, wie Forschende positive Fortschritte machen. Darüber hinaus wird darauf hingewiesen, wo Verbesserungen notwendig sind, um Compliance zu gewährleisten.
Was ist euer Rat für andere nicht-kommerzielle Studiensponsoren, die das Meldeverhalten ihrer klinischen Studien verbessern möchten?
Seid vorbereitet und gewillt, intensive Aktivitäten durchzuführen, um die Einträge zu aktualisieren. Eventuell mithilfe eines engagierten Task Teams, das die Aufgaben sanft und verständnisvoll angeht. Später, das ist sehr entscheidend, sollten klare Rahmenbedingungen gesetzt werden, Erwartungen abgefragt und Anleitungen für die Pflege der Register erarbeitet werden. All dies sollte den Forschenden eindeutig kommuniziert werden, damit Verantwortlichkeiten klar sind und der erzielte Fortschritt stabil bleibt und zur Routine werden kann.
TranspariMED dankt der Universität Nottingham, dass sie ihre Erfahrungen mit der breiteren medizinischen Forschungsgemeinschaft teilt. Diese Fallstudie ist auch einzeln als PDF Download verfügbar in der Sammlung von Transparenzinstrumenten, die TranspariMED für Universitäten zusammengestellt hat: www.transparimed.org/resources
Der vorliegende Text wurde ursprünglich von Till Bruckner verfasst, von Hannah Eger übersetzt und von Jörg Schaaber überarbeitet. Er unterliegt wie das Original [5] einer Creative Commons BY 3.0 Lizenz.
Artikel aus dem Pharma-Brief 4-5/2019, S.10
Bild Universität Nottingham © dietrichherlan/istock
[1] Medicines and Healthcare products Regulatory Agency
[2] Clinical trials of investigative medicinal products
[3] www.who.int/topics/clinical_trials/en
[4] DeAngelis et al. (2004) Clinical trial registration: a statement from the International Committee of Medical Journal Editors. Lancet; 364, p 911
[5] www.bukopharma.de/images/pressemitteilungen/2019/Clinical_Trial_Transparency_EU_Universities_2019.pdf
WHA: Deutschland auf Distanz zu Transparenz-Beschluss
Bundesregierung verteidigt undurchsichtige Strukturen im Pharma-Markt
Die Geheimniskrämerei bei Medikamentenpreisen und klinischen Studien schadet PatientInnen weltweit. Sie behindert einen fairen Zugang zu Arzneimitteln ebenso wie den Zugang zu verlässlichen Informationen. Am 28. Mai hat die Weltgesundheitsversammlung (WHA) eine Resolution angenommen, die dem entgegenwirken möchte. Der deutlich ambitioniertere, ursprüngliche Entwurf ist jedoch massiv aufgeweicht worden. Deutschland blockierte besonders energisch und übte heftige Kritik am Verfahren.
Ein Schritt in die richtige Richtung – so sahen es Viele nach Abschluss des tagelangen Ringens in Genf. WHO-Generaldirektor Dr. Thedros Adhanom Gheyebresus sprach von einem Meilenstein.[1] Vor allem Länder und zivilgesellschaftliche Organisationen des globalen Südens aber auch beachtlich viele aus dem Norden hatten schon früh Unterstützung für die Initiative signalisiert.
Die Resolution, ursprünglich von Italien eingebracht, umfasst mehrere Handlungsfelder.[2] Beispielsweise sollen die tatsächlich gezahlten Medikamentenpreise weltweit bekannt gemacht werden. Bislang sind die mit einzelnen Ländern ausgehandelten Rabatte meist geheim. Außerdem soll öffentlich werden, wo die Hersteller für neue hochpreisige Medikamente gar keine nationale Zulassung beantragen, weil der Markt zu wenig Profit verspricht. Auch die Ergebnisse von klinischen Studien sollen ausnahmslos veröffentlicht werden. Nicht zuletzt will die Resolution für mehr Transparenz bei Forschungskosten und -finanzierung sorgen. Allerdings wurde der Ursprungstext gerade bei diesem Thema stark verwässert. In der verabschiedeten Fassung heißt es daher nur noch: „Die 72. Weltgesundheitsversammlung […] stellt die Wichtigkeit der Finanzierung der Forschung und Entwicklung von Gesundheitsprodukten durch den öffentlichen wie privaten Sektor fest, und sucht die Transparenz dieser Finanzierung entlang der Wertschöpfungskette zu verbessern.“[2]
Deutsche Delegation rügt Verfahren
Deutschland war in Genf ein echter Bremsklotz bei der Suche nach ambitionierteren Lösungen. Zunächst beschwerte sich die Delegation über Prozessmängel, da das italienische Gesundheitsministerium die Resolution erst einen Tag nach Sitzung des WHO-Exekutivrates eingereicht hatte. Das Gremium beriet entsprechend nicht über den Text, was unüblich ist, aber in der Vergangenheit durchaus schon vorkam. Zudem wurde der fast viermonatige Vorlauf als zu geringer Zeitrahmen kritisiert.
Zu einem „echten diplomatischen Thriller“ (so der Lancet) waren die Verhandlungen spätestens geworden, als das WHO-Sekretariat versehentlich einen Entwurf mit nationalen Änderungswünschen unabgestimmt veröffentlichte, der natürlich schnell von Medien und NGOs verbreitet wurde.[3] Spätere Änderungen im Resolutionstext waren mit Beginn der WHA dann ohnehin öffentlich. So lässt sich nachverfolgen, dass Deutschland, ungeachtet seines Protests gegen das Verfahren, in den inoffiziellen wie formellen Verhandlungen umfangreiche Umformulierungen und Streichungen einbrachte, um progressive Forderungen zu entkernen oder komplett zu tilgen.[4] Gegen Ende blieb die Delegation dann den Verhandlungen fern und stand schließlich mit Ungarn und Großbritannien allein auf weiter Flur, als die drei Länder sich in einem ungewöhnlichen Schritt entschlossen, offiziell auf Distanz zur finalen Version zu gehen.[5]
Subventionen weiter geheim?
Dass ausgerechnet Deutschland sich derart in die Bresche wirft, um den pharmafreundlichen Status Quo zu wahren, ist erschreckend. Schließlich hat sich die Bundesregierung deutlich zu ihrer Verantwortung im Bereich globale Gesundheit bekannt, beansprucht hier eine Führungsrolle und hat in den letzten Jahren durchaus auch begrüßenswerte Initiativen auf den Weg gebracht hat. Die eingebrachten Änderungswünsche in der aktuellen WHA-Resolution sprechen jedoch eine deutlich andere Sprache.
So torpedierten die Deutschen Bemühungen, die Forschungskosten der Pharma-Hersteller und deren Preisgestaltung transparenter zu machen. Sogar die in den Forschungskosten enthaltenen öffentlichen Zuschüsse und Subventionen sollten nicht aufgedeckt werden. Zum Vergleich: Spanien ging der verabschiedete Resolutionstext beim Thema Forschungskosten nicht weit genug. Außerdem wandte sich Deutschland gegen das Bestreben, mehr Licht in das Patentdickicht zu bringen, um etwa Anfechtungen von fragwürdigen Patenten (Patentopposition) oder Zwangslizenzen zu vereinfachen.
Der breiten Kritik aus der politischen Opposition in Deutschland begegnete das Bundesministerium für Gesundheit mit Unverständnis, wie das Ärzteblatt schreibt: „Die Arzneimittelpreise in Deutschland seien transparent, hieß es vom Ministerium. Allerdings erkenne man die Arzneimittelrabatte, die zwischen Industrie und Krankenkassen ausgehandelt würden, als Geschäftsgeheimnisse an.“[6]
Kritik an der Kritik
Auf der Ziellinie des WHA-Verhandlungsmarathons stand neben den Inhalten der Resolution auch Verfahrenskritik im Vordergrund. Abseits von Deutschland rügten auch andere Staaten wie Australien und Schweden den untypischen Prozess, primär die als zu kurz wahrgenommene Dauer, sowie das Fehlen einer Debatte im Exekutivrat. Brasilien und Spanien widersprachen dieser Auffassung wiederum, nannten den Prozess fair und die Zeit ausreichend.
Die deutsche Seite hingegen sah gar eine „Verletzung des Geistes von Genf“. [7] Sie erhob den Vorwurf, es habe Versuche gegeben, Delegationen „öffentlich einzuschüchtern“. Dabei habe „man unzutreffende Informationen über die Gründe für diese Positionen durchsickern lassen.“[8]
Ellen 't Hoen, Direktorin von Medicines Law & Policy und früher unter anderem aktiv für Health Action International und den Medicines Patent Pool, war in Genf vor Ort und widerspricht dieser Leseart: „Deutschland war von Tag 1 an gegen diese Resolution. Nun verwechselt es demokratische Prozesse, in denen politisch Handelnde zur Rechenschaft gezogen werden und Medien selbstverständlich eine Rolle zukommt, mit einer „Attacke“.[9]
Das größere Bild
Tatsächlich hatte es im Vorfeld der Verabschiedung der Resolution eine ganze Reihe offener Briefe von zivilgesellschaftlichen Organisationen aus dem Süden und Norden gegeben.[10] Auch intensive Social-Media-Kampagnen wurden während der WHA lanciert, besonders via Twitter. Es wäre der bessere Schluss gewesen, hätte Deutschland diese laute öffentliche Kritik als Ausdruck der Dringlichkeit und Reichweite des Themas interpretiert. Etliche Länder haben das getan und trotz anfänglich kritischer Haltung aus freien Stücken für die Resolution gestimmt. Manche betonten dabei ausdrücklich, trotz des vielleicht nicht optimalen Prozesses die inhaltlichen Aussagen der Resolution für sehr wichtig zu halten.
Zivilgesellschaft als böser Bube
Wirft man einen Blick auf aktuelle WHO-Prozesse, so erscheint die rhetorische Schärfe der deutschen Reaktion zudem in einem besonders problematischen Licht. Im Zuge ihrer Umstrukturierung möchte die WHO u.a. die Zusammenarbeit mit zivilgesellschaftlichen Akteuren neu regulieren. Einige Vorschläge dazu sind höchst kritikwürdig, wie mehrere NGOs (darunter auch die Pharma-Kampagne) in einem aktuellen Papier mahnen.[11] Auf der Exekutivratsitzung nach der WHA wurden jene Vorschläge auch diskutiert. Beschwert sich nun Deutschland, wie das in eben jener Sitzung geschah, nachdrücklich über Medien und NGOs, ist dem Diskurs sicher nicht geholfen.[12] Zumal nicht in einer Zeit, in der „schrumpfende Räume“ Zivilgesellschaft und Journalisten weltweit zunehmend zu schaffen machen.
Dünnes Alibi
Abseits der aufgeheizten Atmosphäre lassen sich mit kühlem Blick vor allem zwei Dinge aus den Genfer Chaostagen ablesen. Zum einen, dass die Bundesregierung von der Welt beim Wort genommen wird, wenn sie sich als treibende Kraft für eine bessere globale Gesundheit darstellt. Zumal, wenn dies so öffentlichkeitswirksam geschieht, wie zuletzt etwa bei der Gründung des Global Health Hub Germany (wir berichteten[13]).
Die Bundesregierung sollte diesen von ihr selbst geschürten Erwartungen konstruktiv begegnen und Farbe bekennen. Ihr Rückzug auf Prozessmodalitäten als Hauptgrund für die WHA-Blockade ist stattdessen ein äußerst dünnes Alibi.[14] Ein Blick auf die von Deutschland eingebrachten Änderungswünsche, die die Resolution weitgehend zahnlos gemacht hätten, zeigt dies in aller Klarheit.
Ins Abseits manövriert
Zum anderen ist festzustellen, dass das Thema Transparenz im medizinischen Bereich mit aller Macht in die globale Öffentlichkeit drängt. Die von der Resolution angesprochenen Probleme sind zwar nicht neu, doch erst unter dem Eindruck steigender finanzieller Belastung der Versorgungssysteme und PatientInnen in Industrieländern lässt sich die globale Debatte nicht mehr ausbremsen.
Die Weltgesundheitsorganisation hat mit der Transparenz-Resolution in weiten Bereichen ein klares Mandat erhalten, für mehr Durchblick bei Arzneimittelpreisen, Forschungskosten und Studienergebnissen zu sorgen. Aber auch die Mitgliedsstaaten der WHO müssen ihre Hausaufgaben machen. Die Bundesregierung täte gut daran, sich den Herausforderungen zu stellen, statt vor strukturellen Problemen die Augen zu verschließen. Nicht nur ihre Glaubwürdigkeit als „Global Health Champion“ steht dabei auf dem Spiel, sondern vor allem die Möglichkeit einer besseren Gesundheit für Menschen weltweit. (MK)
Artikel aus dem Pharma-Brief 3/2019, S.1
Foto © WHO/A.Tardy
[1] Health Policy Watch (2019) World Health Assembly Approves Milestone Resolution On Price Transparency. www.healthpolicy-watch.org/world-health-assembly-approves-milestone-resolution-on-price-transparency/ [Zugriff 6.6.2019]
[2] WHO (2019) Improving the transparency of markets for medicines, vaccines, and other health products. http://apps.who.int/gb/ebwha/pdf_files/WHA72/A72_ACONF2Rev1-en.pdf [Zugriff 7.6.2019]
[3] Lancet (2019) UK, Germany, dissociate from WHO drug pricing resolution. 393, p 2287 https://linkinghub.elsevier.com/retrieve/pii/S0140-6736(19)31329-7
[4] WHO (2019) Improving the transparency of markets for medicines, vaccines, and other health products. Entwurf vom 23.5.2019 www.keionline.org/30823 [Zugriff 24.6.2019]
[5] Der bei der WHO verwendete Begriff in diesem Zusammenhang ist „dissociate“.
[6] Ärzteblatt (2019) Streit um WHO-Resolution für mehr Transparenz bei Arzneimittelkosten. www.aerzteblatt.de/nachrichten/103372/Streit-um-WHO-Resolution-fuer-mehr-Transparenz-bei-Arzneimittelkosten [Zugriff 6.6.2019]
[7] Devex (2019) Transparency, migrant health wrap up 72nd World Health Assembly. www.devex.com/news/transparency-migrant-health-wrap-up-72nd-world-health-assembly-95001 [Zugriff 7.6.2019]
[8] Reuters (2019) WHO agrees on watered down resolution on transparency in drug costs. www.reuters.com/article/us-health-pricing/who-agrees-watered-down-resolution-on-transparency-in-drug-costs-idUSKCN1SY0W4 [Zugriff 7.6.2019]
[9] Persönliche Mitteilung vom 29.5.2019. Weitere Informationen zur Arbeit von Medicines Law & Policy: https://medicineslawandpolicy.org/
[10] Siehe http://bit.ly/OpenLetterToGermany [Zugriff 7.6.2019] und www.bukopharma.de/images/aktuelles/Offener_Brief_WHA_72_Transparenz_Resolution.pdf
[11] Geneva Global Health Hub (2019) Towards a more meaningful engagement of WHO with civil society. http://g2h2.org/posts/civilsocietyengagement/ [Zugriff 07.06.2019]
[12] Health Policy Watch (2019) WHO’s EB Considers New Ways To Work With NGOs – Some Countries Criticise Activists’ Role At WHA 72. https://www.healthpolicy-watch.org/whos-eb-considers-new-ways-to-work-with-ngos-some-countries-criticise-activists-role-at-wha72/ [Zugriff 14.06.2019]
[13] Pharma Brief (2019) Interessengruppen auf den Leim gegangen? Nr. 8-9, S. 1
[14] Deutscher Bundestag (2019) Plenarprotokoll 19/103. Anlage 2. Schriftliche Antworten auf Fragen der Fragestunde (Drucksache 19/10536). Frage 48. S. 12591. http://dipbt.bundestag.de/dip21/btp/19/19103.pdf [Zugriff 14.06.2019]
Wege in die Unabhängigkeit
Für bessere Evidenz ohne Industrieeinfluss
Die angesehene britische Fachzeitschrift BMJ unterstützt die globale Initiative “Pathways to independence” von WissenschaftlerInnen, ÄrztInnen, Bewertungsagenturen und Zivilgesellschaft, die den kommerziellen Einfluss bei der Wissensgewinnung und Weitergabe eindämmen will.[1]
Spätestens seit dem bahnbrechenden Bericht des US-Institute of Medicine (IOM) vor zehn Jahren ist das Thema Interessenkonflikte im Gesundheitswesen auf der globalen Agenda. Die neue Initiative versucht, das Thema breiter aufzustellen und vom Ende her zu denken: Welcher Evidenz bedarf es, damit BehandlerInnen und Erkrankte die bestmöglichen Entscheidungen für oder gegen eine Behandlung treffen können.
Interessenkonflikte untergraben nicht
nur die Glaubwürdigkeit der Medizin, sie führen auch zu Überdiagnose und -behandlung, suboptimalen Therapieentscheidungen und tragen zur Verschwendung knapper Ressourcen bei.
Der Appell orientiert sich an den drei vom IOM identifizierten Bereichen der Einflussnahme: Forschung, Ausbildung und Praxis. Allerdings geht er über die ursprüngliche Forderung, Interessenkonflikte transparent zu machen, weit hinaus: Die „endemischen finanziellen Verwicklungen“ müssten weitgehend aufgelöst werden, damit die Versorgung besser werden kann.
Verzerrte Ergebnisse
Global gesehen sind über 60% der Medikamentenforschung durch die Industrie finanziert.[2] „Es wurde wiederholt gezeigt, dass die veröffentlichten Ergebnisse von industriegeförderten Studien die Produkte des Sponsors bevorzugen. Das schafft einen ‚Sponsorship Bias‘ in der Evidenzbasis, der den Nutzen übertreibt und den Schaden herunterspielt.“ [1] Das ist aber längst noch nicht alles: Wenn LeiterInnen von klinischen Studien persönlich Geld von Herstellern erhalten, fallen die berichteten Ergebnisse positiver aus.[3]
Die Verzerrung von Ergebnissen kann auf vielerlei Wegen geschehen. Dazu gehören unfaire Vergleiche, das „Weglassen“ von für den Sponsor unvorteilhaften Ergebnissen oder durch Metaanalysen, die aber die verzerrten Ergebnisse lediglich als nur scheinbar unabhängige Analyse reproduzieren.
Selbst die Zulassungsbehörden, die VerbraucherInnen schützen sollen, leben sowohl in den USA als auch in der EU größtenteils von Gebühren, die sie von der Industrie kassieren. Strengere Prüfungen würden die Zahl der Anträge reduzieren und die Finanzierungsbasis der Behörden gefährden.
Es käme also darauf an, unabhängige Forschung zu fördern, deren Ergebnissen man wirklich vertrauen kann. Das hätte zudem den Vorteil, dass sich die Forschung stärker am medizinischen Bedarf orientieren würde. Finanziert werden könnte öffentliche Forschung zum Teil aus Ersparnissen durch günstigere Produkte (es gibt ja keine Notwendigkeit sie durch hohe Preise zu refinanzieren). Essenziell ist auch die unabhängige Bewertung von Ergebnissen. Cochrane, die größte Institution, die systematische Reviews von Studien macht, hat nach heftiger Kritik[4] ihre Regeln für Interessenkonflikte immerhin verschärft.
Wes‘ Brot ich ess
Die Aus- und Fortbildung von medizinischem Fachpersonal ist stark von der Industrie unterwandert. Dabei kann gezeigt werden, dass es einen direkten Zusammenhang zwischen der Zahl der von einer Firma bezahlten Essen mit der Verschreibung der Produkte des großzügigen Spenders gibt.[5] Hier ist die Forderung eindeutig: Medizinische Fortbildung muss unabhängig sein. Norwegen kann hier als Vorbild dienen: Für gesponserte Veranstaltungen gibt es keine Fortbildungspunkte mehr. Auch einige Fachzeitschriften achten inzwischen darauf, dass Berichte über neue Medikamente nur noch von AutorInnen ohne Interessenkonflikte verfasst werden. Den zweiten Schritt, keine Pharmawerbung mehr zu schalten, sind bislang aber nur wenige gegangen. Eine löbliche Ausnahme sind die in der International Society of Drug Bulletins (ISDB) organisierten Zeitschriften, die schon seit Jahren weder Werbung noch AutorInnen mit Interessenkonflikten akzeptieren.[6]
Mitmachen
Der Appell richtet sich an die Öffentlichkeit und kann mit unterzeichnet werden. Außerdem wird explizit aufgefordert, auf weitere Evidenz zum Thema hinzuweisen und weitere Bereiche zu benennen, die die Evidenzgewinnung behindern oder verzerren. Auch Lösungsvorschläge sind willkommen.[7] (JS)
Artikel aus dem Pharma-Brief 10/2019, S. 5
[1] Moynihan et al. (2019) Pathways to independence: towards producing and using trustworthy evidence. BMJ; 367, p l6576
[2] Moses III H et al. (2015) The Anatomy of Medical Research US and International Comparisons. JAMA; 313, p 174
[3] Pharma-Brief (2017) Wer zahlt, bestimmt die Musik. Nr. 4, S. 6
[4] Pharma-Brief (2018) Kritiker rausgeworfen. Nr. 8-9, S. 7
[5] De Jong C et al. (2016) Pharmaceutical Industry-Sponsored Meals and Physician Prescribing Patterns for Medicare Beneficiaries. JAMA Int Med;176, p 1114
[6] Interessenkonflikt: Der Pharma-Brief ist Mitglied von ISDB
[7] www.bmj.com/commercial-influence
Vom Wert unabhängiger Information
Treffen der unabhängigen Arzneimittelzeitschriften
Alle drei Jahre trifft sich die International Society of Drug Bulletins (ISBD), um über aktuelle Entwicklungen zu diskutieren. Im Oktober war es wieder so weit. Ein Bericht aus Paris, der viele Schwachstellen bei der Medikamentenkontrolle und -Information deutlich machte.
Gleich die erste Podiumsdiskussion unter dem Titel „Das regulatorische Umfeld – wie gute Evidenz brauchen wir?“ verlief äußerst kontrovers.
Während für Jordi Llinares von der europäischen Zulassungsbehörde EMA die Welt weitgehend in Ordnung ist, sahen das die übrigen RednerInnen anders.
Claudia Wild vom Ludwig Bolzmann Institut, das in Österreich für Arzneimittelbewertungen zuständig ist, zeigte am Beispiel von Krebsmedikamenten, dass zum Zeitpunkt der Zulassung viel zu wenig über den Nutzen der Arzneimittel bekannt ist. Und was man weiß, ist nicht gerade ermutigend.[1] Legt man die Kriterien der europäischen Krebsfachgesellschaft an, zeige nur jedes Fünfte bis Zehnte Mittel einen klinisch relevanten Nutzen, so Claudia Wild. Über die Zeit betrachtet werde auch die Studienqualität immer schlechter. Immer häufiger würden Surrogatendpunkte wie progressionsfreies Überleben (PFS) statt das tatsächliche Überleben (OS) gemessen.[2]
ISDB-Präsident Dick Bijl und Sidney Wolfe von Worst Pills – Best Pills aus den USA prangerten die beschämend niedrigen Standards für die Zulassung von Antidiabetika an. Sotagliflozin war von der US-Zulassungsbehörde nur haarscharf nicht als Zusatztherapie bei Diabetes Typ 1 zugelassen worden: die Abstimmung der ExpertInnen endete mit einem Patt. Die EMA gab dem Wirkstoff hingegen im April 2019 grünes Licht, obwohl klar war, dass ein hohes Risiko für eine Ketoazidose – eine bedrohliche Übersäuerung des Blutes – besteht, so Sidney Wolfe. 
Sanofi brachte das Medikament trotz Zulassung zunächst nicht auf den Markt. Im Juni wurden neue Daten bekannt: Sotagliflozin verschlechtert die Nierenfunktion.[3] Am 26. Juli 2019 gab Sanofi das Ende der Zusammenarbeit mit der Firma Lexicon, die den Wirkstoff ursprünglich entwickelt hatte, bekannt. Als Begründung wurden explizit enttäuschende Studienergebnisse bei der Wirksamkeit genannt.[4] Sotagliflozin hatte von der EMA eine ganz normale Zulassung bekommen. Sanofi wurde lediglich auferlegt, eine Studie zur Häufigkeit von Ketoazidosen durchzuführen.
Llinares verteidigte dagegen die beschleunigte vorläufige Zulassung, selbst bei einer schwachen oder unvollständigen Evidenz. Seiner Meinung nach sei das kein Problem, da bislang nur eines der so zugelassenen Mittel wieder vom Markt genommen werden musste (zwei weitere Hersteller zogen „aus kommerziellen Gründen“ ihren Zulassungsantrag zurück). Bei den meisten der Schnellzulassungen ist nach mehreren Jahren immer noch unklar, ob und welchen tatsächlichen Nutzen die Wirkstoffe für PatientInnen haben.
Generell zeigte die Debatte, dass es doch eine erhebliche Kluft zwischen der Denkwelt der Zulassungsbehörde auf der einen Seite und unabhängigen MedikamentenbewerterInnen sowie klinisch tätigen ÄrztInnen auf der anderen Seite gibt. Während sich die Behörde mit statistisch signifikanten Ergebnissen zufrieden gibt, stellen unabhängige ExpertInnen die Frage, ob überhaupt gemessen wurde, was für PatientInnen zählt und ob die Behandlung auch zu relevanten Verbesserungen führt.
Ein weiterer Streitpunkt war die Tatsache, dass die EMA zu rund 90% durch Gebühren der Industrie finanziert wird. Llinares betonte seine Unabhängigkeit, er bekomme sein Gehalt, egal ob er positiv oder negativ entscheide. Was er dabei übersieht: Wenn die EMA strenger urteilen würde, verringerte sich die Zahl der Zulassungsanträge und damit nähmen auch die Einnahmen der Behörde drastisch ab.
Die Kunst des Weglassens
Auch sinnvolle Medikamente richten bei Überverschreibung gravierende Schäden an. Vor allem ältere Menschen nehmen oft eine große Zahl verschiedener Arzneimittel ein, ohne dass das ihrer Gesundheit nützt. Im Gegenteil, die Zahl der unerwünschten Wirkungen und Interaktionen nimmt zu. Es ist aber gar nicht so einfach, Medikamente abzusetzen. Dazu bedarf es der konstruktiven Zusammenarbeit zwischen ÄrztIn und PatientIn, dem Aufgeben liebgewordener Gewohnheiten und es braucht Mut. So reagiert nicht jeder Arzt entspannt darauf, wenn jemand weniger Medikamente schlucken möchte. Dee Mangin berichtete über diese Klippen und wie man sie überwindet. Sie entwickelte in Kanada gemeinsam mit KollegInnen eine Software, die das „Entschreiben“ (De-prescribing, im Gegensatz zu Verschreiben) erleichtert.[5] Anne Sophie Parent von der AGE-Platform ergänzte die Diskussion aus PatientInnensicht.
Wes Brot ich ess
Interessenkonflikte spielen eine immer noch unterschätzte Rolle in der Medizin. Einen erfrischenden Einstieg ins Thema bot Zoé Friedmann von der Berliner Studierendengruppe von UAEM.[6] Sie setzt sich für Unabhängigkeit in der Lehre ein. Es ist ungut, wenn ProfessorInnen in Vorlesungen bestimmte Medikamente in den Vordergrund stellen, ohne ihre Interessenkonflikte offenzulegen. UEAM untersuchte in einer kleinen Studie, wie deutsche Medizinfakultäten mit dem Problem umgehen. Das Ergebnis ist gelinde gesagt ernüchternd (mehr dazu auf S. 2).
Danach ging es um die Unabhängigkeit der Cochrane Collaboration. Im vergangenen Jahr hatte sie sich nicht gerade mit Ruhm bekleckert als sie das Vorstandsmitglied Peter Gøtzsche erst von seinen Aufgaben entband und ihn als Gründungsmitglied dann sogar ganz ausschloß. Sein „Vergehen“: er hatte den laxen Umgang mit Interessenkonflikten kritisiert und daraus resultierende fehlerhafte Analysen von Cochrane kritisiert.[7] Juan Erviti aus Pamplona, einer der Koordinatoren der Cochrane Hypertension Group – eine der Gruppen, die das Thema Interessenkonflikte sehr ernst nimmt – beschrieb den Umgang mit durch Beeinflussung verzerrten Daten. Er berichtete auch, dass der Skandal um Peter Gøtzsche bereits einiges verändert habe. Die Regelungen zu Interessenkonflikten würden künftig verschärft. Aber in der anschließenden Diskussion wurde deutlich, dass das nicht reicht. Das meinte nicht nur Gøtzsche, der in Paris anwesend war, und sein neu gegründetes Institute for Scientific Freedom vorstellte.[8]
Big Data – Big Problems
Gerd Antes, ehemaliger Leiter des deutschen Cochrane-Zentrums beleuchtete in seinem Vortrag die Chancen und Risiken der Auswertung großer Datenmengen aus dem Internet und aus anderen Datensammlungen. Schon der Titel seines Vortrags „Big Data – marketinggetriebener Hype versus wissenschaftsbasierter Fortschritt, wohin gehen wir?“ machte deutlich, dass er große Gefahren sieht.[9] Ausgangspunkt ist der Goldstandard in der Medizin. Als wichtigste Faktoren sieht er bei der Erkenntnisgewinnung die Minimierung von systematischen Fehlern (risk of bias) und die Kontrolle von zufälligen Fehlern. Randomisierte kontrollierte Studien können – wenn sie gut gemacht sind – wichtige Wissensfortschritte generieren. Dabei gibt es allerdings das Problem, dass rund die Hälfte aller Studien nicht veröffentlicht werden. Also ist schon die Gegenwart alles andere als ideal. Doch bringt die Zukunft Fortschritte?
Bereits seit einiger Zeit geistert der unscharfe Begriff „Real World Data“ durch die Fachblätter. Der angebliche Vorteil: Es würden PatientInnen im Behandlungsalltag erfasst, die gefundenen Ergebnisse seien also praxisnäher. Dabei drohen allerdings grundlegende wissenschaftstheoretische Erkenntnisse ohne Not über Bord geworfen zu werden. Die so erfassten Daten sind nämlich von unterschiedlicher Qualität, es gibt keine Kontrollgruppen und durch die Auswahl der Daten können die tatsächlichen Behandlungseffekte grob über- oder unterschätzt werden.
Aktuell ist der Begriff „Big Data“ in aller Munde. Es existieren große Datensätze, die oft zu anderen Zwecken gesammelt wurden, aber möglicherweise gesundheitsrelevante Informationen enthalten. Die populäre Vorstellung, man muss die Daten nur geschickt auswerten, und kommt so zu bahnbrechenden neuen Erkenntnissen, ist aber schlichtweg falsch. Wenn unstrukturierte Daten analysiert werden, besteht die Gefahr, dass so lange ausgewertet werden, bis ein „Ergebnis“ dabei herauskommt. Solche Ergebnisse sind überdies nicht reproduzierbar, weil jede Sekunde neue Daten hinzukommen. Je mehr Daten man analysiert, umso mehr Korrelationen kann man finden, aber Korrelationen sind noch keine Kausalität. Man denke nur an die Zahl der Störche und die Geburtenrate.
Die praktischen Versuche mit Big Data in der Medizin sind bislang ziemlich ernüchternd. Google scheiterte mit seinem Versuch, Grippeepidemien vorherzusagen. Und IBM wollte mit „Watson“ und künstlicher Intelligenz die Krebsdiagnose und ‑behandlung verbessern. Der Erfolg blieb trotz großen Aufwands aus. Was als Geschäftsmodell in der Warenwelt funktioniert, die Vorlieben von InternetnutzerInnen für Produkte zu erkennen und zielgerichtete Werbung zu schalten, lässt sich nicht eben einfach auf Gesundheit übertragen.
Durch immer mehr Daten besteht die Gefahr, nur den sprichwörtlichen Heuhaufen zu vergrößern, nicht aber die Anzahl der Nadeln, die man finden kann. Statt von „Künstlicher Intelligenz“, so Antes, könne man ebenso von „Künstlicher Dummheit“ sprechen. Jüngstes Beispiel seien die Abstürze von zwei Boeing 737 MAX8s, die durch „mitdenkende“ Software ausgelöst wurden.
Trotz all dieser Bedenken propagieren sowohl die EMA wie die FDA die Verwendung von „Real World Evidence“, wie Rita Banzi vom italienischen Mario Negri Institut zeigte. Besonders bei (zu) frühen Zulassungen von Arzneimitteln spielen zunehmend unstrukturierte Daten eine Rolle.
Die dunkle Seite der Medizin
Unerwünschte Arzneimittelwirkungen sind ein Stiefkind der Medizin. PatientInnen möchten sie nicht erleiden, ÄrztInnen wollen nicht daran schuld sein und für die Industrie sind sie umsatzschädlich. Auch die Kontrollbehörden beschäftigen sich nur ungern damit, könnte man ihnen doch vorwerfen, bei der Zulassung nicht genau genug hingeschaut zu haben.
Antidepressiva können langanhaltende Sexualstörungen verursachen. Der britische Psychiater David Healy (RxISK[10] ), sorgte jüngst dafür, dass diese Nebenwirkung Eingang in die Beipackzettel findet. Die Bereitschaft von PatientInnen, ihr Leiden öffentlich zu machen, war die Voraussetzung dafür.
Marine Martin von APESAC in Frankreich[11] berichtete in Paris als Betroffene über das Valproat-Syndrom. Wenn Schwangere das Epilepsiemedikament nehmen, kann ihr Nachwuchs schwer geschädigt werden. Auch hier dauerte es lange, bis die Berichte der Mütter über geschädigte Kinder ernstgenommen wurden.
Die Opioidkrise in den USA thematisierte Adriane Fugh-Berman von der Georgetown University in Washington. 2017 starben in den USA 49.000 Menschen an einer Überdosis, davon hatten 19.000 das Mittel auf Rezept erhalten und häufig auch in der verordneten Dosis eingenommen. Fugh-Berman zeigte, wie die Hersteller, allen voran Purdue, mit massiven Werbekampagnen die Ausweitung der Indikation – die sich ursprünglich auf Schmerzen bei Krebs beschränkte – vorangetrieben haben. Dabei wurde nicht nur auf Anzeigen gesetzt, sondern es wurden auch Meinungsführer in der Ärzteschaft eingekauft. Und das keineswegs nur in den USA. Grünenthal und Mundipharm, die europäische Schwestergesellschaft von Purdue, zahlten dem italienischen Arzt Guido Fanelli über Jahre insgesamt über eine Million €. Er veröffentlichte nicht nur zahlreiche wissenschaftliche Artikel, die die Gefahren von Opioiden verschleierten. Außerdem organisierte er als Strohmann eine Konferenz, bei der die Firmen die ReferentInnen auswählten. Fanelli war auch Ideengeber für ein Gesetz, dass in Italien die Verschreibung von Opioiden bei chronischen Schmerzen erleichterte. Doch seine verdeckten Aktionen flogen auf. Fanelli wurde nicht nur von seinem Posten im Krankenhaus suspendiert, er darf auch bis zum Abschluss des Falls nicht als Arzt arbeiten.[12]
Barbara Mintzes von der Universität Sydney berichtete über ein von Australien und Kanada gefördertes internationales Projekt zur Risikokommunikation. Dabei wurden Sicherheitswarnungen der Behörden Australiens, Kanada, Großbritanniens und der USA über zehn Jahre (2007-2016) verglichen.[13] Insgesamt wurden 1.441 Warnungen zu 680 Medikamenten oder Wirkstoffgruppen gefunden. Erstaunlich gering war die Übereinstimmung: Warnungen in einem Land lösten häufig keine in den anderen Ländern aus. Betrachtet man nur die Warnungen zu den 573 Medikamenten(gruppen), die überall erhältlich waren, wurden nur 40 in allen vier Ländern veröffentlicht, also nicht einmal jede Zehnte.
Fake news
Mit der Berichterstattung über Arzneimittel beschäftigte sich der Vor-
trag von Alan Cassels von der kanadischen Therapeutics Initiative.[14] Er machte deutlich, wie weit der Einfluss der Pharmaindustrie auch in die etablierte Tagespresse hinein reicht. Und damit auch die Klientel der ÄrztInnen und WissenschaftlerInnen erreicht. Bereits seit fast 30 Jahren ist bekannt, dass Fachartikel, über die in der Laienpresse berichtet wird, anschließend von WissenschaftlerInnen häufiger zitiert werden.[15] Auswahlkriterium ist für die JournalistInnen aber nicht unbedingt die Qualität der Studien sondern das erwartete öffentliche Interesse an der Thematik. Statt vorrangig über randomisierte Studien zu berichten, die die zuverlässigsten Ergebnisse liefern können, werden in der Berichterstattung oft fehleranfälligere Beobachtungsstudien aufgegriffen – und dann auch noch methodisch schlechte.[16]
Einen ganz anderen Input lieferte Anna Coretchi vom moldawischen Bulletin MEDEX. Sie hat eine fünfzehnjährige Karriere als TV-Journalistin hinter sich, und ist bis heute mit Medizinthemen im Fernsehen präsent. Sie ermutigte die Bulletins, aktiv auf die Presse zuzugehen.
Arbeit voraus
 Die Tagung zeigte nicht nur wichtige Problembereiche auf, die einer rationalen Anwendung von Medikamenten im Wege stehen, sie diente auch zur Orientierung, wo ISDB in den nächsten drei Jahren Schwerpunkte setzen könnte. Bessere Studien und strengere Zulassungsbedingungen einzufordern, gehört ebenso auf die Agenda von ISDB wie mehr Transparenz bei Studienergebnissen. Auch vernachlässigten Themen wie „weniger verschreiben“ und „mehr auf unerwünschte Wirkungen achten“, dürften künftig sicherlich stärker in den Fokus rücken. (JS)
Die Tagung zeigte nicht nur wichtige Problembereiche auf, die einer rationalen Anwendung von Medikamenten im Wege stehen, sie diente auch zur Orientierung, wo ISDB in den nächsten drei Jahren Schwerpunkte setzen könnte. Bessere Studien und strengere Zulassungsbedingungen einzufordern, gehört ebenso auf die Agenda von ISDB wie mehr Transparenz bei Studienergebnissen. Auch vernachlässigten Themen wie „weniger verschreiben“ und „mehr auf unerwünschte Wirkungen achten“, dürften künftig sicherlich stärker in den Fokus rücken. (JS)
Artikel aus dem Pharma-Brief 7-8/2019, S.3
Bild Tagungsmitglieder © Benoit Marchand
Bild Gebäude © Jörg Schaaber
[1] Grössmann N et al. (2019) Monitoring evidence on overall survival benefits of anticancer drugs approved by the European Medicines Agency between 2009 and 2015. Eur J Cancer; 110, p 1
[2] Kordecka A et al. (2019 Selection of Endpoints in Clinical Trials: Trends in European Marketing Authorization Practice in Oncological Indications. Value in Health; 22, p 884
[3] Müller C (2019) Sotagliflozin– reduziert Blutdruck und Nierenfunktion? DAZ online www.deutsche-apotheker-zeitung.de/news/artikel/2019/06/12/sotagliflozin-reduziert-blutdruck-und-nierenfunktion [Zugriff 2.11.2019]
[4] Sanofi (2019) Sanofi provides update on ZynquistaTM
(sotagliflozin) type 2 diabetes Phase 3 program and collaboration with Lexicon. Press release 26 July www.sanofi.com/en/media-room/press-releases/2019/2019-07-26-22-05-00 [Zugriff 2.11.2019]
[6] Universities Allied for Essential Medicines
[7] Pharma-Brief (2018) Kritiker rausgeworfen. Nr. 8/9, S. 7
[9] Einen Vortrag von Gerd Antes zum Thema von 2018 kann hier angesehen werden: https://cast.itunes.uni-muenchen.de/clips/0U9YuGalbb/vod/high_quality.mp4
[11] www.oacscharity.org/apesac
[12] Galofaro C (2019) ‘Pain League’ allegedly pushed opioids in Italy. AP https://apnews.com/5d085839c808473eaabd0a3b57c485e0
[13] Perry LT et al. (2019) Comparative analysis of medicines safety advisories released by Australia, Canada, the United States, and the United Kingdom. JAMA Internal Medicine; 179, p 982
[14] Moynihan R and Casssels A (2005) Selling sickness. Vancouver: Greystone Books
[15] Phillips DP et al. (1991) Importance of the lay press in the transmission of medical knowledge to the scientific community. NEJM; 325, p 1180
[16] Selvary S et al. (2014) Media Coverage of Medical Journals: Do the Best Articles Make the News. PLOS one; 9, p e85355
Vom gelassenen Umgang meilenweit entfernt
HIV/Aids bleibt eine komplexe Herausforderung
Im Kampf gegen HIV/Aids sind Stigma, Diskriminierung und Kriminalisierung immer noch hohe Hürden. Dies zeigte sich jüngst in Singapur, wo vertrauliche Behandlungsdaten von HIV-positiven PatientInnen an die Öffentlichkeit gelangten.
Datenlecks – seit Monaten beschäftigen sie die deutsche Öffentlichkeit, sei es zu Bundestagsmitgliedern (Bundestags-Leak), Bankengeschäften (Panama Papers) oder Spitzensport (Football Leaks). Wenig Beachtung fand hierzulande allerdings ein Vorfall, der massive Folgen für tausende Betroffene hat.
Ende Januar gab das Gesundheitsministerium von Singapur bekannt, dass persönliche Daten von 5.400 Einheimischen und 8.800 AusländerInnen, die als HIV-positiv getestet wurden, geleakt wurden.[1] Der Datensatz umfasste neben der Diagnose Namen, Adressen, Telefon- und Passnummern sowie teils auch Informationen zu Kontaktpersonen.
Misstrauen und Kriminalisierung
Singapur ist ein konservativ geprägtes Land. Die meisten Menschen mit HIV teilen ihre Diagnose dort nur mit sehr Wenigen. So überrascht es nicht, dass die Menschen, die von dem Leak betroffen waren, in Medienberichten ihre Ängste schilderten, berufliche oder private Probleme zu erleiden.[2] Gleichzeitig fanden sich in der lokalen Presse auch Stimmen, die von starken Ressentiments gegenüber den PatientInnen zeugten.[3] In Singapur ist Sex zwischen Männern durch ein Gesetz aus Kolonialzeiten noch immer verboten. Schätzungsweise die Hälfte der jährlich neu Betroffenen hat sich auf diesem Weg angesteckt.[4] AktivistInnen verweisen darauf, dass dies auch eine direkte Folge der Kriminalisierung ist und versuchen, die alte Regelung vor Gericht zu kippen.
Noch bis 2015 verbot der reiche Stadtstaat HIV-positiven Ausländern komplett die Einreise. Mittlerweile sind Aufenthalte unter drei Monaten möglich. Mutmaßlicher Verursacher des Datenlecks war ein US-Amerikaner, der sich laut Aussage der Behörden illegal in Singapur aufhielt. Der Mann habe in Singapur arbeiten wollen, sei aber schon bei seiner Einreise HIV-positiv gewesen und falle daher per Gesetz unter ein Arbeitsverbot. Laut offiziellen Stellen begann er Blutproben zu fälschen, um einreisen und vor Ort bleiben zu können, ehe ihn die Behörden unter anderem wegen Betruges verurteilten und 2018 abschoben.[5] Über Motive für die Veröffentlichung wurden bislang keine Angaben gemacht. Sein singapurischer Partner, der Mitarbeiter des Gesundheitsministeriums war und Zugang zu den Daten hatte, steht in den kommenden Monaten vor Gericht.
Kein Einzelfall
Singapur ist kein Einzelfall, wenn es um diskriminierende Einreisebestimmungen für Menschen mit HIV geht. Dies zeigen Nachforschungen der Global Database on HIV-Specific Travel and Residence Restrictions und von UNAIDS. Die für Aids zuständige UN-Organisation arbeitet momentan an einem neuen Bericht zu dieser Form der Diskriminierung von HIV-Positiven. Das letzte Datenblatt konstatierte: „Stand Juni 2015 hielten noch 36 Länder, Territorien und Gebiete Einschränkungen der Reisefreiheit aufrecht.“ [6]
Der beschriebene Fall wirft allerdings auch ein grelles Schlaglicht auf ein gravierendes Problem der HIV/Aids-Prävention. Die toxische Verbindung von Stigma, Diskriminierung und Kriminalisierung erschwert die weltweite Aids-Bekämpfung weiterhin massiv. Peter Wiessner, Mitautor der Global Database, stellt zu den Ereignissen in Singapur fest: „Die Basis von Diskriminierung bilden Irrglaube und Angst. Beim Thema HIV geht es dabei um Drogennutzung, Männern die Sex mit Männern haben und all jene Realitäten, die Länder nicht wahrhaben wollen. Xenophobie spielt ebenfalls eine Rolle.“ [7]
Gefährliche Trias
Stigma, Diskriminierung und Kriminalisierung sind trotz aller Erfolge im globalen Kampf gegen HIV/Aids ein hohes Hindernis. Augenfällig ist dies gerade in Regionen, wo die Zahl der Neuinfektionen steigt, beispielsweise in Teilen Osteuropas, Zentralasien und dem Mittleren Osten. Das durch UNAIDS herausgegebene Global Aids Update 2018 trug auch vor diesem Hintergrund den bezeichnenden Titel „Miles to go“. [8]
Diskriminierung beeinträchtigt zudem die Wirksamkeit von NRO-Arbeit im globalen Süden. Hierbei geht es nicht nur um Gesundheitsprojekte im engeren Sinne, sondern auch um Fragen rund um Menschenrechte und Soziales. HIV/Aids ist ein Querschnittsthema und muss als solches begriffen werden. Entsprechend sind Aspekte wie Stigma, Diskriminierung und Kriminalisierung bei der Arbeit zu Gesundheit als Menschenrecht, ebenso wie bei Maßnahmen zur Förderung von Mädchen und Frauen oder Programmen gegen andere Infektionskrankheiten zu berücksichtigen. Nur so lassen sich die noch existierenden sozialen Hindernisse bei der Bekämpfung der Seuche überwinden. (MK)
Artikel aus dem Pharma-Brief 1/2019, S.3
Bild © Eustaquio Santimano
[1] Reuters (2019) U.S. citizen leaks data on 14,200 people in Singapore with HIV. www.reuters.com/article/us-singapore-health/u-s-citizen-leaks-data-on-14200-people-in-singapore-with-hiv-idUSKCN1PM17T [Zugriff 12.02.2019]
[2] Deutsche Welle (2019) Entsetzen in Singapur über geleakte HIV-Daten. www.dw.com/de/entsetzen-in-singapur-%C3%BCber-geleakte-hiv-daten/a-47280174 [Zugriff 13.02.2019]
[3] Medical Xpress (2019) Fury at HIV data leak in conservative Singapore. https://medicalxpress.com/news/2019-02-fury-hiv-leak-singapore.html [Zugriff 12.02.2019]
[4] Ives M (2019) Data breaches dent Singapore´s image as a tech innovator. New York Times 29 Jan www.nytimes.com/2019/01/29/world/asia/singapore-data-breach-hiv.html [Zugriff 12.02.2019]
[5] Washington Post (2019) An American hid his HIV status to survive in Singapore. Exposed, he allegedly punished thousands living with the virus. www.washingtonpost.com/nation/2019/02/01/an-american-hid-his-hiv-status-survive-singapore-exposed-he-punished-thousands-living-with-virus-authorities-say/?utm_term=.81f8301664a7 [Zugriff 12.02.2019]
[6] UNAIDS (2015) Lifting HIV-related restrictions on entry, stay and residence https://open.unaids.org/sites/default/files/documents/FINAL_TR_A3_press.pdf [Zugriff 12.02.2019]
[7] South China Morning Post (2019) VISA restrictions for HIV-positive immigrants still in place in dozens of countries. www.scmp.com/lifestyle/travel-leisure/article/2185009/visa-restrictions-hiv-positive-immigrants-still-place [Zugriff 12.02.2019]
[8] UNAIDS (2018) Global AIDS Update 2018. Miles to go – Closing Gaps. Breaking Barriers. Righting Injustices. www.unaids.org/sites/default/files/media_asset/miles-to-go_en.pdf [Zugriff 12.02.2019]
Unklare Route bei Arzneipreisen
Bundesregierung weicht bei Transparenzfragen aus
Hohe Medikamentenpreise betreffen verstärkt auch den globalen Norden, so ist Bewegung in die weltweite Debatte um Lösungen gekommen. Ein Aspekt dabei ist verstärkte Transparenz bei Kosten und Preisen. Welche Rolle die Bundesregierung einnehmen möchte, bleibt jedoch unklar. Eine Chance läge im konstruktiven Dialog mit der Zivilgesellschaft.
Eine Kleine Anfrage der Bundestagsfraktion der LINKEN vom 14. August setzte sich noch einmal mit den massiven Konflikten um die Transparenzresolution während der Weltgesundheitsversammlung (WHA) in diesem Frühjahr auseinander. Die Bundesregierung wurde zu ihrer damaligen „Dissoziierung“ von der durch Italien eingebrachten Transparenzresolution (wir berichteten[1]) befragt. Die Antwort vom Parlamentarischen Staatssekretär des Bundesgesundheitsministeriums verdeutlicht vor allem, dass ein nachhaltiger Austausch zur deutschen Position bei dem Thema weiterhin dringend vonnöten ist.
Lieber gar nicht, als zu wenig
Wie so oft bei entsprechenden Anfragen bleibt die Rückmeldung in vielen Punkten äußerst vage. Bekräftigt wird zunächst die Kritik am Vorgehen der Italiener, so wäre ein vorangehendes Einbeziehen des Exekutivrates bei der Resolution „angemessen“ gewesen. Auch hätte dies mehr Vorbereitungszeit gegeben.[2] Das hätte allerdings bedeutet, dass das Thema nicht mehr auf die Agenda der diesjährigen WHA gekommen wäre. Diese Strategie hatte die Bundesregierung in Genf verfolgt. Sie wurde aber von den meisten Mitgliedsstaaten nicht geteilt, für die der dringende Handlungsbedarf im Vordergrund stand.
Inhaltlich wird zunächst allgemein festgestellt: „Die Bundesregierung unterstützt das Ziel der Resolution, den Zugang zu Arzneimitteln und anderen Gesundheitsprodukten weltweit zu verbessern.“ Stante pede folgt jedoch die Einschränkung: „Allerdings handelt es sich um ein komplexes Thema. […] Eine ausschließliche Betrachtung von Einzelkomponenten wie z.B. der Preistransparenz wird den Herausforderungen nicht gerecht.“[3]
Entlang dieser Argumentationslinie werden die weiteren Fragen abgehandelt. So sei eine Offenlegung von Daten zu Verkaufszahlen, Kosten klinischer Studien oder Umfang öffentlicher Subventionen „nicht belastbar“ oder könne „zu falschen Rückschlüssen führen“. Der Tenor: lieber gar keine Transparenz, als unvollständige. Zudem wird ein weiteres Mal auf den bundesdeutschen Kontext verwiesen: „Zu der Forderung nach einer Veröffentlichung von Arzneimittelpreisen hat die Bundesregierung erläutert, dass Rabatte, die kassenindividuell mit pharmazeutischen Unternehmen vereinbart werden, als Vertragsbestandteile dem Betriebs- und Geschäftsgeheimnis unterliegen.“ Das trifft zwar auf Rabattverträge für Generika zu, ist aber auch hierzulande wegen der Korruptionsanfälligkeit solcher Vereinbarungen keineswegs unumstritten. Für die viel wichtigere Nutzenbewertung neuer meist hochpreisiger Arzneimittel sind die ausgehandelten Rabatte bekannt. Man kann sie zum Beispiel im Arzneiverordnungs-Report nachlesen.[3]
Inkonsequente Schlüsse
Paradoxerweise stellt die Antwort des Ministeriums zu Recht fest, maßgeblich für PatientInnen sei, „[…] welchen therapeutischen Zusatznutzen ein neues Arzneimittel im Vergleich zur bisherigen Standardtherapie bietet. Die in Entwicklung und Produktion eingeflossenen Kosten treffen darüber keine Aussage. So sollte beispielsweise ein neues Arzneimittel nicht deshalb einen höheren Preis erzielen, weil es mit hohem finanziellen Aufwand entwickelt wurde.“[3] Dass allerdings die Pharmaindustrie eben jene vermeintlich exorbitanten Forschungskosten für Präparate, die aufgrund mangelnder Transparenz nur schwer überprüfbar sind, seit jeher als ein Hauptargument für hohe Arzneimittelpreise ins Feld führt, wird einfach ausgeblendet.
Am Ende der Stellungnahme steht schließlich die Feststellung, die Bundesregierung verfolge einen „holistischen Ansatz zur weltweiten Verbesserung des Zugangs zu Arzneimitteln und anderen Gesundheitsprodukten“, wie er auch in der WHO Access Road Map 2019-2023 vorgesehen sei. In eben jener taucht Transparenz allerdings äußert prominent auf, etwa mit der Empfehlung zur Förderung von Transparenz bei Forschungs- und Entwicklungskosten sowie zur globalen und regionalen Zusammenarbeit für erhöhte Preistransparenz.[4]
Wie hältst Du´s mit der Transparenz?
Die Bundesregierung unterstützt nach eigenem Bekunden einen verbesserten Zugang zu Arzneimitteln, sieht jedoch Transparenz als nur einen Faktor von vielen und hadert mit dessen Komplexität. Bei dieser Gemengelage stellt sich die Frage, was das konkret für das zukünftige Engagement bedeutet.
Wie groß das Bedürfnis nach der Klärung der deutschen Perspektive ist, zeigte zuletzt eine Initiative aus der Zivilgesellschaft. In einem Offenen Brief vom 2. September an Bundesgesundheitsminister Spahn, lanciert vom Aktionsbündnis gegen AIDS und unterzeichnet von elf Nichtregierungsorganisationen, darunter auch der Pharma-Kampagne, wird ein inhaltlicher Dialog angeregt. So heißt es: „Aus unserer Sicht ist […] bislang offen geblieben, wie die Bundesregierung prinzipiell zum Inhalt der Transparenz-Resolution steht und inwiefern sie abweichende Positionen vertritt. […] Ein früher und offener Dialog hierzu kann für die weitere Arbeit zu den gesundheitsrelevanten SDGs wichtige und positive Impulse setzen.“ [5]
Auch angesichts des zerschlagenen Porzellans auf der letzten WHA bedarf es nun gemeinsamer Anstrengungen, denn klar ist: Die Gretchenfrage, wie es die Bundesregierung mit der Preistransparenz bei Medikamenten, Impfstoffen und Gesundheitsprodukten wirklich hält, wird sich in den kommenden Jahren wieder und wieder stellen.
Ein umfassender Zugang zu Gesundheitsversorgung weltweit ist ohne bezahlbare Produkte undenkbar. Ohne verbesserte Transparenz wird der Kampf gegen Preistreiberei aber nicht erfolgreich sein können. Untätig zu bleiben ist angesichts der krassen globalen Versorgungslücken keine Option.
Im Bereich Globale Gesundheit richtet sich die Aufmerksamkeit besonders auf das UN High-Level Meeting zu Universal Health Coverage (UHC), das am 23. September – nach Redaktionsschluss dieser Ausgabe – stattfand. Bereits in der Vorbereitung des Treffens sorgte das Thema Transparenz abermals für Zündstoff.[6] (MK)
Artikel aus dem Pharma-Brief 6/2019, S.3
Bild © Jörg Schaaber
[1] Pharma Brief (2019) WHA: Deutschland auf Distanz zu Transparenz-Beschluss. Nr. 3, S. 1
[2] BMG (2019) Antwort auf Kleine Anfrage der Abgeordneten Sylvia Gabelmann, Susanne Ferschl, Matthias W. Birkwald und weiterer Abgeordneter und der Fraktion DIE LINKE. betreffend „Deutschland in den Verhandlungen zur Transparenzresolution“, BT-Drs 19/12382. https://sylvia-gabelmann.de/wp-content/uploads/2019/09/2019-08-28-AW-PSt-Dr.-Gebhart_KA-19_12382.pdf [Zugriff 12.09.2019]
[3] Schwabe et al. (2018) Arzneiverordnung-Report 2018. Heidelberg/Berlin: Springer
[4] WHO (2019) Draft Road Map for Access to medicines, Vaccines and other health products, 2019–2023. https://apps.who.int/gb/ebwha/pdf_files/WHA72/A72_17-en.pdf [Zugriff 12.09.2019]
[5] Aktionsbündnis gegen Aids u.a. (2019) Deutschlands Prioritäten bei Zugang und Transparenz im Bereich Globale Gesundheit. www.aids-kampagne.de/sites/default/files/brief_an_bundesgesundheitsministrer_spahn_-_transparenz_resolution_-_19-02-09.pdf [Zugriff 11.09.2019]
[6] Branigan D (2019) Drug R&D, Sexual & Reproductive Health Scrutinised In Draft UHC Declaration. Health Policy Watch, 19 July www.healthpolicy-watch.org/drug-rd-sexual-reproductive-health-scrutinised-in-draft-uhc-declaration [Zugriff 23.07.2019]
Transparenzverlust droht
US-Behörde will weniger über Arzneimittel preisgeben
Bislang dokumentiert die US-Zulassungsbehörde FDA ihren Entscheidungsprozess über neue Medikamente ausführlich. Damit soll künftig Schluss sein. Unter dem Schlagwort „Modernisierung“ [1] soll es nur noch eine Bewertung „light“ geben.
Umfangreiche Berichte über zulassungsrelevante Analysen der FDA-MitarbeiterInnen sind auf der Website der FDA bisher frei zugänglich. Damit stehen ausführliche Bewertungen der vom Hersteller eingereichten Daten zur Verfügung. Da die Studien zum Zeitpunkt der Zulassung oft noch nicht oder nur sehr selektiv publiziert wurden, bieten die FDA-Unterlagen wichtige Informationen für eine unabhängige Einschätzung des Nutzens von Arzneimitteln.
Zweifelhafte Verschlankung
Im Rahmen des “New Drugs Regulatory Program Mode” der FDA sollen die umfänglichen Dokumente künftig durch eine kurze Zusammenfassung der FDA-Bewertung ersetzt werden. Angestoßen hat die „Modernisierung“ der von Präsident Trump vorgeschlagene und nach einigen Querelen bestätigte FDA-Kommissar Scott Gottlieb. Das neue Konzept orientiert sich an der eher bescheidenen Transparenz der europäischen Arzneimittelbehörde EMA. Der sogenannte EPAR[2] enthält nur eine stark komprimierte Fassung des internen Bewertungsberichts der Behörde. Das geschieht in großzügiger Auslegung der EU-Verordnung zur Arzneimittelzulassung, die eigentlich nur die Schwärzung von Geschäftsgeheimnissen vorsieht.[3]
Während die EMA – wegen öffentlichen Drucks – seit einigen Jahren die Transparenz erhöht, vor allem durch die zusätzliche Veröffentlichung der Clinical Study Reports, die die EU-Verordnung zu klinischen Studien vorschreibt, schlagen die USA also die entgegengesetzte Richtung ein.
Substanzverlust
Stellungnahmen von WissenschaftlerInnen, Fachjournalen und NGOs warnen eindringlich vor dem Vorhaben der FDA.[4] Während bislang die einzelnen Abteilungen der FDA unterschiedliche Aspekte der Zulassungsunterlagen separat bearbeiten, alle relevanten Daten transparent dargestellt und auch unterschiedliche Meinungen festgehalten werden, soll nicht nur der öffentliche Zugang zu diesen Unterlagen abgeschafft werden. Auch der Bearbeitungsprozess soll „effizienter“ werden, so Gottlieb im Juni 2018.[5] Das heißt, nicht nur nach außen, auch behördenintern soll es eine integrierte Bewertung geben. Auch wenn der FDA-Kommissar betonte, dass der „Fortschritt in unseren eigenen Arbeitsprozessen durch modernere und stringentere wissenschaftliche Ansätze erreicht werden“ soll, sieht das Vorhaben nach einer Verflachung und Gleichschaltung der wissenschaftlichen Debatte aus.
Darüber versprach Gottlieb, dass externe Interessengruppen – darunter auch die Industrie – in Zukunft „stärker in den wissenschaftlichen Austausch“ während des gesamten Zulassungsprozesses einbezogen werden.
In der Eingabe von Peter Doshi und 35 weiteren WissenschaftlerInnen wird auch auf einen weiteren Widerspruch aufmerksam gemacht.[6] Die Abteilung für Medikamentenbewertung der FDA (CDER) hatte 2010 ein Papier verfasst, das ausdrücklich die Protokollierung von dissenten Meinungen der BearbeiterInnen vorschreibt.[7] Vorgesetzte sind verpflichtet, die Gründe für ihre abweichende Entscheidungen zu protokollieren.
Insgesamt riecht die geplante „Modernisierung“ stark nach einem Wohlfühlprogramm für Big Pharma. Der US-Lobbyverband PhRMA begrüßt denn auch die geplanten Regeln.[8] Allerdings ist er strikt dagegen, dass die FDA ihr Pilotprogramm zur Veröffentlichung von Clinical Study Reports fortsetzt. Dabei wird die wenig begründete Sorge vorgeschoben, dass dadurch krankheitsbezogene Daten von einzelnen PatientInnen identifizierbar würden. Ausgerechnet die EU-Datenschutzverordnung wird von PhRMA ins Feld geführt, obwohl – siehe oben – die EMA inzwischen die Clinical Study Reports veröffentlicht.
Die Industrie hat gleich noch eine längere Wunschliste angehängt. Sie möchte, dass auch Daten aus „real world evidence“, also Anwendungsbeobachtungen, Register u.a. sowie Verbesserungen bei Biomarkern in Zulassungsentscheidungen einfließen.
Gottlieb, der den ganzen Änderungsprozess angestoßen hat, verließ die FDA im April diesen Jahres, angeblich um mehr Zeit für seine Familie zu haben. Seit Juni sitzt er im Direktorium des Pharmamultis Pfizer.[9]
Artikel aus dem Pharma-Brief 6/2019, S.1
Bild FDA Building 51 houses the Center for Drug Evaluation and Research © U.S.-Regierung
[1] Das Programm trägt den Namen “New Drugs Regulatory Program Modernization” www.fda.gov/drugs/regulatory-science-research-and-education/modernizing-fdas-new-drugs-regulatory-program [Zugriff 12.9.2019]
[2] European Public Assessment Report
[3] Der Text der EU-Verordnung 726/2004 lautet: „Die Agentur veröffentlicht – nach Streichung aller vertraulichen Angaben geschäftlicher Art – umgehend den vom Ausschuss für Humanarzneimittel erstellten Bericht über die Beurteilung des Humanarzneimittels und die Gründe für das Gutachten zugunsten der Erteilung einer Genehmigung“
[4] Beschreibung der Einladung zu Kommentaren zum Clinical Data Summary Report Pilot program und alle Eingaben: www.regulations.gov/document?D=FDA-2019-N-2012-0001 [Zugriff 12.9.2019]
[5] Gottlieb S (2018) Statement from FDA Commissioner Scott Gottlieb, M.D., on proposed modernization of FDA’s drug review office. 4 June www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-proposed-modernization-fdas-drug-review-office [Zugriff 12.9.2019]
[6] www.regulations.gov/document?D=FDA-2019-N-2012-0010 [Zugriff 12.9.2019]
[7] FDA (2010) Equal Voice: Discipline and Organizational Component Collaboration in Scientific and/or Regulatory Decisions. 16 Sept. www.fda.gov/media/79353/download
[8] www.regulations.gov/document?D=FDA-2019-N-2012-0022 [Zugriff 12.9.2019]
[9] Feinstein C (2019) Ex-FDA chief Scott Gottlieb just joined the board of $240 billion drugmaker Pfizer. Business Insider, 28 June www.businessinsider.de/scott-gottlieb-goes-from-fda-commissioner-to-pfizer-board-member-2019-6?r=US&IR=T [Zugriff 12.9.2019]
Schlampig gemacht
Wie gut sind Zulassungsstudien für Krebsmedikamente?
Wenn Medikamente gegen Krebs auf den Markt kommen, ist oft noch nicht bekannt, ob sie tatsächlich das Leben der PatientInnen verlängern. Jetzt ging eine AutorInnengruppe noch einen Schritt weiter: Sie überprüfte, was die Studien, die Hersteller bei der europäischen Behörde EMA einreichen, methodisch taugen.[1] Das Ergebnis ist ernüchternd.
Viele Krebsmittel werden auf Basis von Kriterien wie progressionsfreie Zeit oder Ansprechrate zugelassen. Dahinter steht die Annahme, diese Surrogatparameter könnten Auskunft über einen (noch nicht) nachgewiesenen Überlebensvorteil bieten. Jahre nach der Zulassung bestätigt sich die Hoffnung oft nicht, wir berichteten.[2],[3]
Ein aktuelles Beispiel ist Venetoclax gegen multiples Myelom. Bei Auswertung der BELLINI-Studie hatten die PatientInnen doppelt so lang kein Tumorwachstum (22,4 versus 11,5 Monate), aber es waren doppelt so viele tot (21,1% versus 11,3%). Zwei Krebsforscher – einer davon war an der Studie selbst beteiligt – warnen: „Die Ergebnisse dieser Studie lehren uns eine wichtige Lektion über die Grenzen von Surrogatendpunkten, die Konsequenzen für die gesamte Onkologie haben, weit über das multiple Myelom hinaus.“ [4]
Qualität nimmt ab
Die AutorInnen um Huseyin Naci nahmen die 32 Zulassungen für Krebsmedikamente durch die EMA 2014-2016 genauer unter die Lupe. Es wurden dabei Angaben sowohl aus den Studienveröffentlichungen als auch den europäischen Bewertungsberichten (EPAR) der EMA berücksichtigt.
Erstes Ergebnis: Während von 2009-2013 noch 90% der Zulassungsstudien randomisiert waren, also dem höchsten Evidenzstandard entsprachen, traf das in der aktuellen Untersuchung nur noch für 75% zu. Immer mehr Krebsmedikamente werden auf Basis von einarmigen Studien – also ohne direkten Vergleich zugelassen. Aber selbst wenn es einen Vergleichsarm gab, war die gewählte Therapie öfters zweifelhaft.
Die Hersteller hatten 41 randomisierte Studien [5] eingereicht, nur bei diesen kann ein Vorteil gegenüber dem bisherigen Therapiestandard überhaupt zuverlässig erkannt werden. Bei 39 lagen genügend Informationen für eine Auswertung vor. Nur zehn Studien (26%) hatten Gesamtüberleben als primären Endpunkt, die übrigen Surrogatendpunkte.
Hälfte schwächelt
Bei 49% der Studien wurden schwerwiegende Schwächen beim Design, der Durchführung oder der Auswertung gefunden. Dabei waren Studien, die Surrogatendpunkte maßen, zu über der Hälfte (55%) methodisch schwach, Studien, die das Überleben maßen, dagegen wesentlich seltener (20%).
Viele Mängel in der Studiengestaltung und –durchführung fanden sich nur im EPAR wieder, aber nicht in den Artikeln, die in Fachzeitschriften veröffentlicht wurden. Dabei war aber auch die Informationstiefe im EPAR sehr unterschiedlich. Oftmals fehlten aussagekräftige Angaben auch dort.
Am häufigsten fehlten bei der Auswertung der Daten eine erhebliche Zahl der PatientInnen. Das stellt die Glaubwürdigkeit in Frage, insbesondere wenn die Ergebnisse der Studienarme nicht weit auseinanderliegen. Bei Nivolumab fehlten die Daten für 16,5% der Personen im Versuchsarm, aber nur weniger als 1% im Vergleichsarm. Es wurden auch keine Sensitivitätsanalysen durchgeführt, indem verschiedene Daten für die fehlende Werte eingesetzt wurden. Das ist ein etabliertes Verfahren, um etwas sicherer zu sein, ob die Lücken nicht zu einer Fehleinschätzung des Nutzens führen.
Geblinzelt
Nicht alle Studien waren verblindet (weder PatientIn noch BehandlerIn wissen welches der geprüften Medikamente gegeben wurde). Das kann zu Überschätzungen des Erfolgs führen: Bei Trametanib wurde die progressionsfreie Zeit sowohl vom behandelnden Personal als auch von verblindeten ExpertInnen bewertet. Erstere ermittelten ein Hazard Ratio (HR[6]) von 0,39 also einen deutlicheren Vorteil, die verblindeten nur 0,55. In der Publikation fand sich nur der günstigere Wert. Auf vollständige Verblindung wird oft wohl verzichtet, weil das kostengünstiger ist – man spart so teure Fachleute.
Generell wird kritisch angemerkt, dass die Lebensqualität in drei Viertel der Studien nicht gemessen wurde. Da die Medikamente häufig in einer palliativen Situation gegeben werden, also die Überlebensdauer der PatientInnen begrenzt ist, ein schweres Versäumnis. (JS)
Artikel aus dem Pharma-Brief 6/2019, S.5
[1] Naci H et al. (2019) Design characteristics, risk of bias, and reporting of randomised controlled trials supporting approvals of cancer drugs by European Medicines Agency, 2014-16: cross sectional analysis. BMJ; 366, p l5221
[2] Pharma-Brief (2017) Bescheidener Fortschritt. Nr. 8-9, S. 1
[3] Pharma-Brief (2017) Viel Lärm um nichts? Nr. 4, S. 4
[4] Shaji Kumar and Vincent Rajkumar SV (2019) Surrogate endpoints in randomised controlled trials: a reality check. Lancet; 394, p 281
[5] Die übrigen Studien waren nicht randomisiert (2) oder hatten gar keinen Vergleichsarm (11)
[6] Ein Hazard Ratio von 1 bedeutet, dass es keinen Unterschied gibt, also das Ereignis in beiden Gruppen gleich häufig eintritt. Kleinere Wert als 1 bedeuten einen Vorteil, höhere Werte einen Nachteil.
Neues aus dem Nebel
BMG-Netzwerk bleibt riskantes Provisorium
Der Global Health Hub Germany (GHHG) ist auch nach seinem Start noch eine massive Baustelle. Zugleich bestätigen neue Einblicke, dass ganz grundlegende Fragen zum Projekt in den Hintergrund geschoben werden.
Zweieinhalb Monate nach der offiziellen Eröffnung des GHHG durch das Bundesministerium für Gesundheit (BMG) bleibt das Projekt ein Stein des Anstoßes. Die Chance, der zivilgesellschaftlichen Kritik[1] durch klare Bekenntnisse zu begegnen, wurde auf politischer Seite mehrfach verpasst. So wurden in einer kleinen Anfrage der Bundestagsfraktion der LINKEN vom 18. März viele neuralgische Punkte des Hubs angesprochen. Und auch auf einer öffentlichen Ministeriumsveranstaltung am 12. April sollte der GHHG Thema sein. In beiden Fällen allerdings enttäuschten die Rückmeldungen.
Die Katze im Sack
Mit einem umfangreichen Fragenkatalog hatte Mitte März die LINKE im Bundestag mit einer kleinen Anfrage zum Hub nachgehakt.[2] Darin wird nach den Ideengebern für den GHHG gefragt.[3] Dazu heißt es in der Antwort des BMG: „Vertreter und Vertreterinnen verschiedener nichtstaatlicher Akteursgruppen sind mit dem Wunsch nach mehr Vernetzung und Austausch an das Bundeministerium für Gesundheit (BMG) herangetreten.“ Wer diese Gruppen sind, wird wohlweislich nicht mitgeteilt. Dass vor allem die Wirtschaft großes Interesse an dem konkreten Projekt hatte, ist jedoch bekannt.1 Wenig erhellend wirken in diesem Zusammenhang auch die Äußerungen der Gesellschaft für Internationale Zusammenarbeit (GIZ), die die Aufgabe hat, den Hub zu koordinieren. Auf die Kritik an der einseitigen Genese des GHHG reagierte die GIZ im Lancet ausweichend.[4] Fakt bleibt, dass die Ausrichtung des Hubs im Wesentlichen schon festgelegt war, ehe die Zivilgesellschaft in die Diskussion mit einbezogen wurde.
Laut dem BMG soll der Lenkungskreis, dem eine elementare Rolle im GHHG zukommt, aus rund 16 Mitgliedern bestehen – zwei aus jeder vom Ministerium berücksichtigten Akteursgruppe. Wie befürchtet, wird der erste Kreis jedoch nicht gewählt, sondern von Ministeriumsseite bestimmt. Nach welchem Prozedere dieses Gremium, das den Großteil der Richtungsentscheidungen treffen soll, dann arbeitet, ist nach wie vor völlig unbekannt. „Einzelheiten einer Verfahrensordnung werden gegenwärtig diskutiert“, heißt es dazu lapidar. Die für eine Mitgliedschaft Umworbenen bekommen momentan also die sprichwörtliche Katze im Sack angeboten.
Keine langfristige Finanzierung
Auf die Frage, wie unabhängig der Hub sei, ist die Antwort in dem Bundestagsdokument interessanter, als es auf den ersten Blick erscheinen mag: „Da der Hub in den ersten drei Jahren durch Mittel der Bundesregierung finanziert ist, ist er vom Einfluss privater Geber und der Gesundheitsindustrie unabhängig.“ Zum einen bezieht sich diese Aussage ausschließlich auf die Anschubphase, der Hub ist jedoch als Projekt ohne begrenzte Laufzeit angelegt. Ob die Politik auch nach den drei Jahren noch sämtliche Kosten trägt, bleibt offen. Würden Mitglieder anschließend bei der Finanzierung eingespannt werden, würde dies schnell zu Lasten ressourcenschwächerer Akteure gehen und das Standing von Industrie und Stiftungen weiter aufwerten. Laut der GIZ, die das Sekretariat des Hubs stellt, soll noch ein nachhaltiges Finanzierungskonzept für die Zukunft erarbeitet werden.4 Zum anderen stellt sich die Frage, welche Hub-Gruppierungen am ehesten die Möglichkeit besitzen, thematische Arbeitsgruppen (AGs) innerhalb des GHHG ins Leben zu rufen und langfristig zu bespielen. Die AGs sind abseits des Lenkungskreises tätig und sollen auch konkrete Projektideen behandeln. Hier sind finanzielle und personelle Ressourcen ebenfalls zentral und bei einigen Akteursgruppen (etwa Jugend und Zivilgesellschaft) deutlich limitiert.
Pfusch am Bau
Noch dürftiger fiel die Debatte des Themas in einer öffentlichen Veranstaltung zur globalen Gesundheitspolitik aus, zu dem das BMG im Berliner GIZ-Sitz nicht-staatliche Akteure geladen hatte. Das Update zum Hub fiel darin in den Schlussbeitrag, zu dem in der Agenda seltsamerweise keine Fragerunde eingeplant war. Echte Neuigkeiten gab es letztlich nur wenige. Laut BMG hat der Hub bislang fast 170 Mitglieder, Details wurden nicht bekannt. Als erste Aktivität soll ein so genannter Global Health Talk nun Mitte Juni stattfinden und den Startschuss für AGs markieren.
In Anbetracht der vielen ungeklärten Punkte ist dieses Vorgehen verwunderlich. Ein Aphorismus, dem Komponisten Anton Bruckner zugeschrieben, besagt: „Wer hohe Türme bauen will, muss lange am Fundament verweilen.“ Im Falle des GHHG, der auch international ein Zeichen für Deutschlands Engagement für globale Gesundheit setzen soll, hat das BMG diese Sorgfalt bislang definitiv nicht walten lassen. Augenblicklich spricht wenig dafür, dass sich dies noch ändert. (MK)
Artikel aus dem Pharma-Brief 2/2019, S. 4
[1] Pharma-Brief (2018) Abgekartetes Spiel. Nr. 8-9, S. 1
[2] Deutscher Bundestag (2019) Drucksache 19/8479. Kleine Anfrage der Abgeordneten Eva-Maria Schreiber u.a. und der Fraktion DIE LINKE. http://dip21.bundestag.de/dip21/btd/19/084/1908479.pdf [Zugriff 08.04.2019]
[3] Deutscher Bundestag (2019) Drucksache 19/9164. Antwort der Bundesregierung auf die Kleine Anfrage der Abgeordneten Eva-Maria Schreiber u.a. und der Fraktion DIE LINKE. http://dip21.bundestag.de/dip21/btd/19/091/1909164.pdf [Zugriff 15.04.2019]
[4] Green A (2019) Germany´s Global Health hub. Lancet; 393, p 862
Neue Perspektiven
Jahresbericht 2018 der BUKO Pharma-Kampagne
Ob Diabetes, vernachlässigte Krankheiten oder Klimawandel – trotz massiver Engpässe im Budget hat die Pharma-Kampagne im vergangenen Jahr eine Vielzahl an Themen bearbeitet, globale Gesundheitsprobleme aus neuer Perspektive betrachtet und daraus Handlungsempfehlungen abgeleitet. Ein Rückblick auf unsere Bildungs- und Advocacyarbeit im Jahr 2018.
Ein besonderer Fokus lag 2018 auf den Folgen des Klimawandels für die globale Gesundheit. Mit unserer Kampagne „Globale Gesundheit braucht Klimaschutz!“ haben wir die aktuelle Debatte um eine wirksame und nachhaltige Klimapolitik befeuert und dabei deutlich gemacht: Investitionen in den Klimaschutz sind immer auch ein Plus für die Gesundheit weltweit. Sie machen sich darum doppelt bezahlt.
Wir haben eine Posterserie und Online-Informationen zum Thema erstellt, aber auch eine Unterrichtseinheit für Berufsschulen sowie einen Pharma-Brief Spezial – pünktlich zur Weltklimakonferenz in Polen. Nicht zuletzt brachte die diesjährige Theatertournee das Thema mit bissigem Humor auf die Straße: Bei 43 Auftritten in 13 Städten sahen sich etwa 2.600 ZuschauerInnen das Stück an, darunter über 800 Schülerinnen und Schüler an Berufsschulen. Die Vorstellungen wurden dort durch eine interaktive Einführung und moderierte Unterrichtsdiskussionen ergänzt. Ein 7-minütiger Film zur Theatertournee steht online. Er zeigt Ausschnitte aus dem Stück und fängt Stimmen von ZuschauerInnen und Mitwirkenden ein.
Online Tool Diabetes
Zum Welt-Diabetes-Tag am 14. November ging außerdem unser neuer E-Learning-Kurs zu Diabetes online. Gerade Länder im globalen Süden sind von der chronischen Erkrankung besonders betroffen. Rund 80% der erwachsenen Menschen mit Diabetes leben in Ländern mit niedrigem und mittleren Einkommen. Doch die deutsche Entwicklungszusammenarbeit wird dieser Herausforderung bisher noch nicht gerecht. Unser Kurs zur globalen Diabetesproblematik soll hier Abhilfe schaffen, umfassend informieren, Handlungsoptionen aufzeigen und zu einer besseren Versorgung der PatientInnen in armen Ländern beitragen.
Sozial gerechte Patentverwertung
In unserem Projekt zur sozial gerechten Patentverwertung haben wir einen Leitfaden zur sozialverträglichen Patentverwertung erstellt und als Pharma-Brief Spezial gemeinsam mit Ärzte ohne Grenzen, Brot für die Welt und der DAHW Deutsche Lepra- und Tuberkulosehilfe veröffentlicht. Das internationale Netzwerk Health Action International brachte die Broschüre zusätzlich in englischer Sprache heraus. Das Heft wurde auch an zahlreiche ExpertInnen für Technologietransfer verschickt und bei öffentlichen Veranstaltungen vorgestellt.
Die Publikation soll alternativen Lizenzverträgen den Weg ebnen, damit innovative Produkte öffentlicher Forschung wie etwa Medikamente oder Impfungen in armen Ländern zu günstigen Preisen verfügbar werden.
Pressearbeit
Schließlich haben wir mit unserer intensiven Presse- und Öffentlichkeitsarbeit Themen gesetzt und dabei immer wieder auch die deutsche Arzneimittel- und Gesundheitspolitik aus internationaler und globaler Perspektive bewertet. Die Pharma-Kampagne war bei 75 Veranstaltungen, Tagungen, Fachgesprächen und Konferenzen im In- und Ausland vertreten, häufig mit Vorträgen, dezidierten Stellungnahmen oder auf dem Podium. Das Spektrum reichte von Unterrichtsbesuchen und Uni-Seminaren bis hin zur Teilnahme an Veranstaltungen im Europäischen Parlament oder Konsultationsprozessen des Gesundheitsministeriums.
Inhaltliche Schwerpunkte waren z.B. der Handlungsbedarf bei Tuberkulose und Antibiotikaresistenzen, vernachlässigte Krankheiten, alternative Forschungsmodelle sowie die globale Gesundheitsstrategie der Bundesregierung und der geplante Global Health Hub Germany. Unsere dezidierte Kritik an dem fragwürdigen Diskussionsforum, das auf eine Initiative der Industrie-Lobby zurückgeht und in dem die Gates-Stiftung mitmischt, wurde breit kommuniziert und floss u.a. in ein Briefingpapier von Brot für die Welt ein.[1]
Wir publizierten acht Pharma-Briefe und beleuchteten in unserer Berichterstattung wunde Punkte der deutschen, europäischen sowie internationalen Gesundheits- und Arzneimittelpolitik. Unethische Arzneimittelstudien in Peru standen dabei ebenso im Fokus wie Interessenkonflikte bei der Weltgesundheitsorganisation, das neue EU-Forschungsprogramm oder die Nutzenbewertung von Arzneimitteln in der EU. Wir thematisierten offensichtliche Schwächen bei der Arzneimittelkontrolle bzw. -zulassung im Fall von krebserregenden Valsartan-Präparaten oder dem fragwürdigen Brustkrebspräparat Palbociclib.
Außerdem standen wir 30 JournalistInnen Rede und Antwort, führten Interviews und Hintergrund-Gespräche oder vermittelten Kontakte zu ExpertInnen im In- und Ausland. Insgesamt gingen 58 Medienberichte aus unserer Pressearbeit hervor. Themenschwerpunkte waren Klimawandel, Arzneimittelpreise, nicht-übertragbare Krankheiten, Antibiotikaresistenzen oder der Einfluss von Gates auf die WHO. Unter anderem berichteten der Spiegel, Frontal 21, Report Mainz, WDR, Hessischer Rundfunk und Deutschlandradio, aber auch das Bundesgesundheitsblatt sowie die Fachzeitschriften E&Z oder Dr. med. Mabuse.
Nicht zuletzt hat auch unsere Webseite im vergangenen Jahr ein neues Gesicht bekommen – besuchen Sie uns doch auf www.bukopharma.de und erfahren Sie mehr über unsere Kampagnen und Projekte! (CJ)
Artikel aus dem Pharma-Brief 1/2019, S. 6
Bild © Jörg Schaaber
[1] Yee-Sol Chang und Karolin Seitz (2019) SDG 3 – Deutschlands Engagement für Globale Gesundheit. Briefing Paper Brot für die Welt/GFP vom Januar 2019 www.brot-fuer-die-welt.de/fileadmin/mediapool/2_Downloads/Fachinformationen/Sonstiges/Briefing_0119_SDG3_online.pdf [Zugriff 6.2.19]
Musterknabe auf Abwegen
Ringen um Zugang zu neuen Impfstoffen
Die Coalition for Epidemic Preparedness Innovations (CEPI) sollte durch innovative Impfstoffforschung neue Wege in der Epidemie-Bekämpfung gehen. Dafür gab es auch umfangreiche Förderung aus Deutschland. Nun steckt das junge Projekt in einer hausgemachten Krise, ausgelöst durch die Verwässerung seiner Zugangspolitik.
Medizinische Innovation ohne angemessenen Zugang ist so problematisch wie häufig. Besonders fragwürdig wird es, wenn eine Neuentwicklung mit öffentlichen Geldern finanziert wurde und PatientInnen am Ende dennoch mit schlechter Verfügbarkeit konfrontiert sind. Ein aktuelles Beispiel dafür ist die hohe Preishürde für das Präparat Truvada® (Emtricitabin/Tenofovir) in den USA. Nationale Forschungseinrichtungen, fast ausschließlich finanziert durch Steuergelder, hatten einen neuen Anwendungsbereich von Truvada erforscht und eine Therapie zur HIV-Präexpositionsprophylaxe (PrEP) entwickelt. Das Medikament kann nun präventiv eingesetzt werden, um stark gefährdete Personen aus Hochrisikogruppen vor einer HIV-Infektion zu schützen. Der Hersteller Gilead hält jedoch trotz des neuen Anwendungsbereichs an seinem alten Produkt-Patent fest und diktiert in den USA den Preis. Dieser liegt bei 1.600 bis 2.000 US-Dollar monatlich.[1] In Europa gibt es hingegen inzwischen viel günstigere Generika.[2]
Ein globaler Versicherungsschein
Dass öffentliche Forschungsinvestitionen am Ende die Kassen privater Unternehmen füllen, ist kein Einzelfall. Daher spielt bei der Vergabe öffentlicher Mittel die Ausgestaltung sogenannter Access Policies eine elementare Rolle. Sie können über verbindliche Regelungen den breiten Zugang zu Forschungsergebnissen sichern. Welches Konfliktpotenzial dieses Thema bietet, zeigt sich momentan bei der der Coalition for Epidemic Preparedness Innovations (CEPI).
2017 – noch unter dem Eindruck des vorangegangenen Ebola-Ausbruchs in Westafrika – gegründet, sollte die Koalition eine schnellere medizinische Reaktion auf Pandemien ermöglichen. Der Fokus lag auf der Impfstoffentwicklung gegen vernachlässigte Krankheiten, zunächst primär gegen MERS-CoV (Middle East respiratory syndrom-related coronavirus), Lassa-Fieber und das Nipah-Virus. Die öffentlich-private Partnerschaft, bestehend aus nationalen Regierungen, der EU-Kommission, der WHO, Forschungsinstitutionen, Wirtschaftsakteuren, Organisationen der Zivilgesellschaft sowie privater und öffentlicher Stifter sollte nach eigenen Worten ein „globaler Versicherungsschein“ sein.[3]
CEPI stellt einen speziellen Fall in der Impfstoffforschung dar. Zum einen aufgrund seines gigantischen Fördervolumens von 1 Mrd. US-Dollar für die erste Förderphase. Zum anderen durch den großen Umfang der Leistungen. So werden nicht nur Forschungsaktivitäten weitreichend finanziert, sondern auch die Arzneimittelproduktion zur Bevorratung neuer Präparate. Hinzu kommen vielfältige Beratungsleistungen für die Partner.
Protest und halbgare Antworten
Die ursprünglichen CEPI-Regelungen zum Zugang bei den entwickelten Produkten und kreierten Daten waren recht umfangreich und fortschrittlich.[4] Ende 2018 allerdings beschloss das Board der Koalition grundlegende Änderungen. Das Ergebnis war eine massive Aufweichung der Leitlinien hin zu einer lapidaren Grundsatzerklärung und ein Schritt zu deutlich mehr Intransparenz.[5]
Infolge dessen schrieben Ärzte ohne Grenzen (MSF), die an der Gründung der Koalition beteiligt waren und sich mit Nachdruck gegen die Neuregelung gewandt hatten, einen offenen Brief an den Verwaltungsrat. Darin ist auch erwähnt, dass die Änderungen anscheinend auf Beschwerden aus der Industrie zurück zu führen sind.[6] Die NGO sähe nun „keine Grundlage mehr dafür, dass CEPI gegenüber seinen öffentlichen und philanthropischen Gebern eine Rechenschaftspflicht über die Vereinbarungen mit Forschungspartnern an den Besitzrechten von geistigem Eigentum, den Umgang mit geistigen Eigentumsrechten oder die Preisgestaltung von CEPI finanzierten Impfstoffen hat.“[5] Auch andere zivilgesellschaftliche Akteure kritisierten, dass Absichtserklärungen in Sachen Access ohne Unterfütterung durch konkrete und transparente Prozesse wertlos seien.[7]
Nach zunächst wenig ergiebigen Antworten seitens CEPI folgte schließlich ein achtseitiges Dokument, u.a. zum Umgang mit Partnerverträgen, Vermarktung und Datentransparenz.[8] Doch auch dabei fielen einige Punkte schnell negativ ins Auge. So tritt CEPI quasi die gesamten Rechte an geistigem Eigentum, wohlgemerkt entwickelt durch öffentliche und private Förderung, schon von Vornherein an private Vertragspartner ab.
Welche Position Deutschland als wichtiger Förderer (100 Mio. US-Dollar) in dieser Auseinandersetzung einnehmen wird, scheint augenblicklich unklar. Als Repräsentant sitzt das Bundeministerium für Bildung und Forschung im Board von CEPI. Klar ist jedoch, eine Partnerschaft, die gerade auch den Verwundbarsten helfen soll, darf sich nicht auf dünne Grundsatzerklärungen verlassen. Anders ausgedrückt: Ein bezahlter „Versicherungsschein“ muss auch gedeckt sein. (MK)
Artikel aus dem Pharma-Brief 2/2019, S. 1
[1] Rowland C (2019) An HIV treatment cost taxpayers millions. The government patented it. But a pharma giant is making billions. Washington Post 26 March https://wapo.st/2FBCXHP?tid=ss_mail&utm_term=.2ba21f2ce73b [Zugriff 15.04.2019]
[2] Ab rund 50 € pro Monat. at-Datenbank [Zugriff 15.4.2019]
[3] Reuters (2017) Global coalition aims to outpace epidemics with new vaccines www.reuters.com/article/us-davos-meeting-vaccines/global-coalition-aims-to-outpace-epidemics-with-new-vaccines-idUSKBN15231Y [09.04.2019]
[4] CEPI (2017) CEPI Policy Documentation https://msfaccess.org/sites/default/files/2018-09/CEPIoriginalPolicy_2017.pdf [09.04.2019]
[5] CEPI (2018) CEPI´s Equitable Access Policy https://cepi.net/wp-content/uploads/2019/01/CEPI-Approach-to-Equitable-Access-13-12-FINAL_0.pdf [Zugriff 09.04.2019]
[6] Ärzte ohne Grenzen (2019) offener Brief zu CEPIs “Equitable Access Policy“. 5. März www.aerzte-ohne-grenzen.de/sites/germany/files/2019-medikamentenkampagne-offener_brief_cepi_board_equitable_access.pdf [Zugriff 09.04.2019]
[7] Devex (2019) Battle over CEPI’s access to vaccines policy deepens. www.devex.com/news/battle-over-cepi-s-access-to-vaccines-policy-deepens-94438 [Zugriff 10.04.2019]
[8] CEPI (2019) Advancing Equitable Access to epidemic vaccines through CEPI’s vaccine and platform development agreements https://cepi.net/wp-content/uploads/2019/03/Advancing-Equitable-Access_CEPI_29032019.pdf [Zugriff 10.04.2019]
Mühseliger Fortschritt
Neuer Tuberkulose-Report zeigt Lücken
Seit 1997 wirft der Global TB Report der WHO jährlich einen Blick auf die weltweiten Dynamiken bei Tuberkulose (TB). Für das zurück liegende Jahr sieht er einige positive Tendenzen. Doch den globalen Bemühungen fehlt es weiterhin an Nachdruck.
Mitte Oktober veröffentlichte die Weltgesundheitsorganisation (WHO) ihren jüngsten Bericht zum Stand der weltweiten Tuberkulosebekämpfung. Vieles in den präsentierten Zahlen liest sich positiv: 7 Millionen Menschen und damit so viele wie nie zuvor erhielten 2018 eine Behandlung gegen TB. Die Anzahl an gemeldeten Todesfällen ging zurück, ebenso die Rate neu gefundener Erkrankungen.[1]
Die Kehrseite ist allerdings: Arme PatientInnen werden durch Eigenleistungen oft ruiniert, resistente TB-Stränge sind weiter ein drängendes Problem und nur ein Drittel dieser neu gemeldeten Infektionen werden behandelt.
Finanzierung dringend gesucht
Eine Damoklesschwert stellen die ebenso chronischen wie massiven Finanzierungslücken im Kampf gegen TB dar. Obwohl eine der tödlichsten Infektionskrankheiten weltweit, bleiben die für TB zur Verfügung gestellten Ressourcen deutlich hinter dem Bedarf zurück. Und neues Ungemach droht: Ein Bericht von Ärzte ohne Grenzen warnte jüngst abermals davor, dass gerade bei Maßnahmen gegen HIV/Aids und TB durch Umstrukturierungen der Förderlandschaft eine verstärkte finanzielle Belastung vieler betroffener Länder erfolgen wird, die diese nicht werden stemmen können.[2]
Selbst das im Oktober erzielte Rekordergebnis der Wiederauffüllungskonferenz des Globalen Fonds gegen Aids, Tuberkulose und Malaria (über 14 Mrd. US-Dollar für die kommenden drei Jahre) kann da berechtigte Sorgen nicht zerstreuen. Der Fonds ist mit Abstand der wichtigste Finanzierer von Programmen zur TB-Diagnostik und Behandlung. Allerdings mahnen kritische Stimmen an, dass das 2014 geänderte Vergabeverfahren des Fonds trotz Nachbesserungen für einige Länder erhebliche Einbußen bedeutet, besonders in Osteuropa.[3] Auch der Verteilungsschlüssel für die drei Krankheiten im Fonds ist umstritten und wird wohl ein kritisches Thema für den nächsten Finanzierungszyklus werden.[4]
Nicht gut, aber besser
Ein wenig besser sieht es mittlerweile in der Pipeline bei Forschung und Entwicklung aus. Es werden mehr Ressourcen investiert und die Pipeline ist heute umfangreicher bestückt als noch vor wenigen Jahren.[5]
Doch die einzigen drei wirklich neuen Präparate der letzten Dekaden stehen wegen hoher Preise und Zulassungslücken immer noch nur einem Bruchteil der PatientInnen zur Verfügung. Bedaquilin und Delamanid haben in der EU nur eine Zulassung unter Auflagen,[6],[7] weil die Daten zur Wirksamkeit und Sicherheit unzureichend sind, Pretomanid ist in Europa noch gar nicht zugelassen. Die Hersteller von Bedaquilin (Johnson & Johnson) und Delamanid (Otsuka) sehen sich daher schon lange massiver Kritik ausgesetzt. Preislich steht das mit öffentlichen Mitteln erforschte Pretomanid (TB Alliance) besser da, doch auch hier sind die Behandlungskosten für ärmere Länder eine signifikante Bürde.[8]
Hoffnung wird mittlerweile wieder in einen Impfstoff gesetzt. Jüngst wurden die endgültigen Daten zu einem Impfstoffkandidaten veröffentlicht. Die vom PDP AREAS und GSK geförderte Studie zeigte immerhin eine Halbierung des Auftretens neuer TB-Fälle. Das ist generell für einen Impfstoff keine gute Quote, aber besser als alles, was bisherige TB-Impfstoffkandidaten an Wirkung erzielten. Weitere Untersuchungen sind nötig, um die Ergebnisse zu bestätigen.[9] Doch selbst bei positivem weiteren Verlauf, würde eine Marktreife erst in einigen Jahren realistisch sein.
Zehrende Zeiten
In den Nachhaltigen Entwicklungszielen der UN (SDGs) findet sich die Selbstverpflichtung, bis 2030 das Ende von TB zu erreichen. Im Zuge ihres neuen Berichtes hebt die WHO dabei die Bedeutung von Universal Health Coverage (UHC) hervor. So einleuchtend der Zusammenhang ist, wird das UHC-Konzept allein keinen Durchbruch bringen können. Denn, so zeigt es auch der Global TB Report 2019, unverändert bilden soziale Determinanten den wohl wichtigsten Faktor in der Bekämpfung. Als die acht Staaten mit der höchsten TB-Bürde weist die WHO für 2018 Bangladesch, China, Indien, Indonesien, Nigeria, Pakistan, die Philippinen und Südafrika aus. Weiterhin sind global vor allem Länder niedrigen Einkommens sowie marginalisierte Bevölkerungsgruppen betroffen. Der Bekämpfung der Armut kommt also eine hohe Priorität zu.
Das erste WHO High-Level Meeting zu Tuberkulose weckte 2018 große Erwartungen. Doch trotz umfangreicher Bemühungen der Zivilgesellschaft fanden nur wenige Staatsoberhäupter überhaupt den Weg nach New York, auch die finale Deklaration ließ viele AktivistInnen ernüchtert zurück.[10] Ein Jahr später bleibt der Eindruck, dass die globalen Bemühungen gegen TB weiter auf einen Art Befreiungsschlag warten. (MK)
Artikel aus dem Pharma-Brief 7-8/2019, S.1
[1] WHO (2019) Global TB Report 2019. https://apps.who.int/iris/bitstream/handle/10665/329368/9789241565714-eng.pdf?ua=1 [Zugriff 04.11.2019]
[2] MSF (2019) Burden sharing or burden shifting? How the HIV/TB response is being derailed. www.msf.org/burden-sharing-or-burden-shifting [Zugriff 04.11.2019]
[3] OSF (2017) Lost in Transition: Three Case Studies of Global Fund Withdrawal in South Eastern Europe. www.opensocietyfoundations.org/publications/lost-transition [Zugriff 04.11.2019]
[4] DEVEX (2019) New tools, old problems: TB funding gap persists. https://www.devex.com/news/new-tools-old-problems-tb-funding-gap-persists-95854 [Zugriff 07.11.2019]
[5] Policy Cures Research (2018) G-Finder 2018. www.policycuresresearch.org/wp-content/uploads/Y11_G-FINDER_Full_report_Reaching_new_heights.pdf
[6] www.ema.europa.eu/en/medicines/human/EPAR/sirturo
[7] www.ema.europa.eu/en/medicines/human/EPAR/deltyba
[8] Reuters (2019) New tuberculosis treatment for developing countries to cost $1,040. www.reuters.com/article/tb-alliance-tuberculosis/corrected-new-tuberculosis-treatment-for-developing-countries-to-cost-1040-idUSL3N27A3R1 [Zugriff 04.11.2019]
[9] Tait DR et al. (2019) Final Analysis of a Trial of M72/AS01E Vaccine to Prevent Tuberculosis. NEJM. http://dx.doi.org/10.1056/NEJMoa1909953
[10] Cousins S (2019) UN High-Level Meeting to end tuberculosis disappointing. Lancet; 392, p 1183 http://dx.doi.org/10.1016/S0140-6736(18)32458-9
Misshandelt bei der Entbindung
Studie deckt Missstände auf
Weltweit werden Frauen bei der Geburt ihrer Kinder misshandelt oder diskriminiert. Eine aktuelle Studie aus mehreren Ländern liefert erschütternde Hinweise. Doch auch in Deutschland ist eine Debatte um Patientinnenrechte längst überfällig.
Meghan Bohren und ihr Team untersuchten Geburtspraktiken in vier Ländern geringen und mittleren Einkommens. Sie beobachteten über 2.000 Geburten in jeweils drei Einrichtung in Ghana, Guinea, Nigeria und Myanmar. Zusätzlich wurden fast 2.700 Frauen in den Tagen nach der Entbindung befragt. Die Ergebnisse sind erschreckend: Mehr als ein Drittel der Mütter hat rund um die Entbindung physische oder verbale Misshandlungen sowie Stigma und Diskriminierung durch die behandelnden ÄrztInnen und PflegerInnen erfahren.
Viele Frauen erlebten vor allem in den letzten15 Minuten der Wehen, kurz vor der Geburt ihres Kindes Gewaltanwendung durch ÄrztInnen und anderes medizinisches Personal. Die Häufigkeit der Misshandlungen variierte stark von Land zu Land, die meisten Fälle traten in Nigeria auf. Auffällig häufig waren insbesondere junge Frauen mit einem geringen Bildungsgrad von Misshandlungen betroffen.
Auch die Privatsphäre der Gebärenden wurde häufig verletzt. In einem Drittel der untersuchten Geburten fehlten Vorhänge oder Trennwände während vaginaler und anderer Untersuchungen. Darüber hinaus wurden medizinische Eingriffe häufig ohne das Einverständnis der Frauen vorgenommen. Über 10% der im Rahmen der untersuchten Geburten durchgeführten Kaiserschnitte und 56% (Umfrage) bzw. 75% (Beobachtungen) der durchgeführten Dammschnitte erfolgten ohne Einwilligung der Patientinnen.[1]
Studien weisen darauf hin, dass die Opfer solchen Missbrauchs Gesundheitsangebote seltener in Anspruch nehmen.[2] Hier wird einmal mehr deutlich, dass es nicht ausreicht, bloß den Zugang zu Gesundheitsversorgung und Behandlung sicherzustellen. Ganz im Sinne von Universal Health Coverage muss es immer auch um die Qualität der angebotenen Dienstleistungen gehen, nämlich darum, „sicherzustellen, dass die Qualität der Versorgungsleistungen gut genug ist, um die Gesundheit derjenigen zu verbessern, die sie in Anspruch nehmen.“ [3] Klare Regeln und Strukturen, die die Rechte von Patientinnen schützen und eine respektvolle und sichere Behandlung garantieren, sind unumgänglich.
Und in Deutschland?
Doch auch hierzulande ist eine Debatte über Gewalt im Kreißsaal längst überfällig. Viele Mütter sprechen inzwischen offen über ihre traumatischen Erfahrungen bei der Geburt und prangern die Missstände an: Ärztliche Willkür, überflüssige schmerzhafte oder gar verletzende Eingriffe wie z.B. ein nicht notwendiger Dammschnitt ohne Einwilligung der Patientin. Die Soziologin Christina Mundlos hat Statistiken ausgewertet und Interviews geführt – zu finden in ihrem Buch „Gewalt unter der Geburt“. Sie geht davon aus, dass rund 50% aller Frauen im Kreißsaal Erfahrungen mit verbalen oder körperlichen Übergriffen macht.[4] Auch wenn die Schätzungen für Deutschland nicht direkt mit den Zahlen aus der Studie von Bohren und KollegInnen verglichen werden können, ein Warnsignal sind sie allemal. (MB/CJ)
Artikel aus dem Pharma-Brief 10/2019, S. 6
[1] Bohren M et al. (2019) How women are treated during facility-based childbirth in four countries: a cross-sectional study with labour observations and community-based surveys.Lancet https://doi.org/10.1016/S0140-6736(19)31992-0 [Zugriff 30.10.2019]
[2] Bohren M et al. (2015) The Mistreatment of Women during Childbirth in Health Facilities Globally: A Mixed-Methods Systematic Review. PLoS Med; 12, p e1001849 https://doi.org/10.1371/journal.pmed.1001847 [Zugriff 30.10.2019]
[3] WHO (2019) Key Facts Universal Health Coverage www.who.int/news-room/fact-sheets/detail/universal-health-coverage-(uhc) [Zugriff 7.11.2019]
[4] Zit. N. M. Haaf (2018) Fass mich nicht an. Süddeutsche Zeitung 5.5. www.sueddeutsche.de/leben/geburtshilfe-fass-mich-nicht-an-1.3930451 [Zugriff 7.11.2019]
Memento Preis 2019
Forschung zu Fieber bei Kindern in Afrika ausgezeichnet
Der Memento Preis für vernachlässigte Krankheiten wurde am 20.2.2019 zum sechsten Mal vergeben. Die diesjährigen PreisträgerInnen sind Prof. Jürgen May (Wissenschaft) und Katharina Nickoleit (Journalismus).
Der Memento Forschungspreis für vernachlässigte Krankheiten ging dieses Jahr an Prof. Dr. Jürgen May vom Bernhard-Nocht-Institut für Tropenmedizin. Die Jury würdigte damit das Engagement des Wissenschaftlers für die Forschung zu Fieberkrankheiten in Afrika. „Die Arbeiten von Professor Jürgen May leisten einen wichtigen Beitrag, Patienten eine effektivere Behandlung zu ermöglichen“, sagte Jurymitglied Prof. Dr. August Stich, Chefarzt der Tropenmedizinischen Abteilung der Missioklinik Würzburg. „Oft wird mangels adäquater Tests irrtümlich von einer Malaria ausgegangen, und die Menschen werden falsch behandelt. Dank Jürgen May und seiner Arbeitsgruppe kann in Zukunft besser festgestellt werden, woran die Menschen tatsächlich erkrankt sind. Das Forscherteam identifizierte häufige Erreger bakterieller Infektionen und entwickelte Strategien zu ihrer besseren Diagnose und Therapie.“
Den Memento Preis in der Kategorie Journalismus erhielt die freie Journalistin Katharina Nickoleit. Mithilfe des Recherchestipendiums möchte sie einen Beitrag über die „Schnell einsetzbare Expertengruppe bei Gesundheitsgefährdungen“ (SEEG) realisieren. Die SEEG wurde von der Bundesregierung, dem Robert-Koch-Institut und dem Bernhard-Nocht-Institut für Tropenmedizin gegründet und kommt dort zum Einsatz, wo eine Krankheit ausbricht, die sich zu einer Epidemie ausweiten könnte.
Das Memento-Bündnis
Ziel der Initiatoren des Preises – Ärzte ohne Grenzen, Brot für die Welt, BUKO Pharma-Kampagne und DAHW Deutsche Lepra- und Tuberkulosehilfe e.V. – ist es, Aufmerksamkeit für vernachlässigte und armutsassoziierte Krankheiten zu schaffen, an denen zwar Millionen Menschen weltweit leiden, für die es aber oft keine adäquaten Impfstoffe, Diagnostika oder Medikamente gibt. Das Memento-Bündnis sieht auch die Bundesregierung in der Pflicht, entsprechend ihrer neuen Führungsrolle in der Globalen Gesundheit mehr Geld für die Forschung im Bereich der vernachlässigten Krankheiten bereitzustellen.
Bild © Jörg Schaaber
Artikel aus dem Pharma-Brief 1/2019, S. 7
Läuft wie geschmiert…
Indien: Antibiotika-Hersteller locken mit teuren Geschenken
Pharmafirmen belohnen indische ÄrztInnen mit Geschenken, wenn sie viele Antibiotika verschreiben. Das britische Bureau of Investigative Journalism[1] deckte diese Werbepraktiken gemeinsam mit dem indischen Journalisten Rahul Meesaraganda auf. Der folgende Artikel ist eine gekürzte und leicht bearbeitete Fassung des englischen Originaltextes von Madlen Davies und Ben Stockton.[2]
 Recherchen des britischen Bureau of Investigative Journalism zufolge belohnen die Firmen Abbott und Sun Pharma in Indien die Verschreibung von Antibiotika mit Geschenken. Zielgruppe der Werbemaßnahmen sind vor allem informelle ÄrztInnen ohne anerkannte medizinische Ausbildung. Diese selbsternannten Doktoren behandeln überwiegend arme PatientInnen, die keinen Zugang zum öffentlichen Gesundheitssystem haben.
Recherchen des britischen Bureau of Investigative Journalism zufolge belohnen die Firmen Abbott und Sun Pharma in Indien die Verschreibung von Antibiotika mit Geschenken. Zielgruppe der Werbemaßnahmen sind vor allem informelle ÄrztInnen ohne anerkannte medizinische Ausbildung. Diese selbsternannten Doktoren behandeln überwiegend arme PatientInnen, die keinen Zugang zum öffentlichen Gesundheitssystem haben.
In den meisten indischen Bundesstaaten ist es zwar illegal, Antibiotika an nicht-
registrierte ÄrztInnen zu verkaufen. Dem Gesetz wird jedoch nur selten Geltung verschafft. Außerdem existiert kein Verbot, diese Medikamente bei informellen ÄrztInnen zu bewerben.
Nicht-registrierte ÄrztInnen praktizieren vor allem in abgelegenen ländlichen Regionen Indiens, aber auch in den Slums der Großstädte. Häufig sind sie vor Ort die einzigen Anbieter von Gesundheitsdienstleistungen und der überwiegende Teil der Bevölkerung ist auf ihre Dienste angewiesen. Doch gerade informelle ÄrztInnen tragen durch häufige Fehlverschreibungen und den exzessiven Gebrauch von Antibiotika in besonderem Maß zur Resistenzbildung bei. Resistente Erreger sind indes ein immenses Problem: Laut Schätzung des Lancet sterben in Indien rund 57.000 Säuglinge jährlich an einer Sepsis, die durch resistente Bakterien verursacht wurde.[3]
Werbegeschenke
Ein Sun Pharma Vertreter berichtete, dass sowohl informellen als auch registrierten ÄrztInnen hochwertige Geschenke gemacht würden, damit sie nicht zur Konkurrenzfirma wechseln. Die Firma sponsert Geschenkgutscheine, medizinische Geräte, Kühlschränke, Fernseher oder Reisen und bietet auch Bargeld oder kostenlose Medikamente als Anreiz, mehr Antibiotika zu verschreiben.
Sun Pharma ist der größte Arzneimittelproduzent in Indien, mit einem Umsatz von über 3,5 Mrd. € in 2018. Abbott India ist die zweitgrößte pharmazeutische Firma im Lande. Laut einem Vertreter von Abbott werden die ÄrztInnen mit Geschenken im Wert von bis zu 2.000 Rupien (25 €) geködert – in Indien eine Menge Geld. Die Firma bot außerdem „Kaufe fünf, bekomme eins gratis“-Deals für Antibiotika-Packungen an, um so Großeinkäufe zu fördern. In einem Land, in dem ÄrztInnen auch selbst Medikamente an die PatientInnen verkaufen dürfen, ein erheblicher Anreiz.
Indiens nicht-zertifizierte Ärztinnen verdienen so wenig, dass diese Anreize ihr monatliches Einkommen bis zu einem Viertel steigern können. Abbott bietet den ÄrztInnen zusätzlich ein Gefühl von Luxus mit Überraschungspartys für ihre Familien und Cocktail Soireen in fünf-Sterne Hotels. Offiziell lassen jedoch beide Firmen verlauten, dass ihre Policies derlei Werbepraktiken verbieten.
ExpertInnen und WissenschaftlerInnen kritisierten gegenüber dem Bureau of Investigative Journalism die Praktiken vehement. Lord Jim O’Neill, der ein Review zur globalen Resistenzproblematik durchgeführt hat, sagte: „Viele Pharmafirmen rühmen sich gerne, dass sie verantwortlich handeln und hier haben wir eindeutige Beweise, die das Gegenteil belegen.“
Professor Ramanan Laxminarayan, Direktor des Centre for Disease Dynamics, Economics and Policy (CDDEP), kommentierte: „Wenn man bedenkt, dass es in Indien fünfmal so viele informelle wie registrierte ÄrztInnen gibt, ist es nicht verwunderlich, dass durch sie die meisten Antibiotika an PatientInnen abgegeben werden. Es ist auch keine Überraschung, dass Pharmafirmen den Verkauf von Antibiotika ankurbeln. Notwendig ist ein Balanceakt, denn einerseits bieten diese informell Praktizierenden den Zugang zu Antibiotika, andererseits muss Über- und Fehlgebrauch vermieden werden. Genau darin liegt die Herausforderung.“
Krise des Gesundheitssystems
Indien ist das Epizentrum der Antibiotikaresistenz-Krise. MedizinerInnen warnen, dass sie regelmäßig PatientInnen mit „pan-resistenten“ Infektionen sehen, resistent gegenüber allen verfügbaren Medikamenten. Gleichzeitig kämpft das Land mit dem fehlenden Zugang zur Versorgung. Theoretisch steht armen PatientInnen in Indien eine kostenlose Gesundheitsversorgung zu. Doch im staatlichen Gesundheitssystem gibt es einen eklatanten Mangel an Fachpersonal. Ein aktueller Bericht des CDDEP ermittelte ein Defizit von 600.000 ÄrztInnen und zwei Millionen KrankenpflegerInnen im öffentlichen Sektor.
Problem informelle ÄrztInnen
Viele Menschen sind deshalb auf die mehr als 2,5 Millionen nicht-registrierten ÄrztInnen angewiesen. Sie praktizieren z.B. traditionelle Heilkunst, Ayurveda, Homöopathie oder Naturheilkunde und sind den rund eine Million privaten oder staatlichen ÄrztInnen mit einer anerkannten wissenschaftsbasierten medizinischen Ausbildung zahlenmäßig bei weitem überlegen.
Oft als „Onkel“ oder „chotto doctor“ (kleiner Doktor) bezeichnet, werden die informellen ÄrztInnen in ihren Gemeinden äußerst respektiert und auch zu allen Alltagsproblemen um Rat gefragt. Sie bieten ein wenig medizinische Versorgung in Regionen, die sonst komplett ohne Gesundheitsversorgung wären.
Dennoch sterben noch immer jedes Jahr mehr als eine halbe Million InderInnen an behandelbaren Infektionen, weil sie keinen Zugang zu Gesundheitsversorgung oder lebensrettenden Arzneimitteln haben.
Geschenke erhalten die Freundschaft
Pharmavertreter von Sun Pharma und Abbott, die mit einem verdeckten Bureau-Ermittler sprachen, machten kein Geheimnis daraus, dass sie sowohl nicht-zertifizierten als auch anerkannten ÄrztInnen Anreize anbieten. Weil Abbott in der Vergangenheit wegen teurer Geschenke für ÄrztInnen stark kritisiert wurde, hat die Firma Einschränkungen eingeführt – aber die Mitarbeiterinnen umgehen diese Regeln. Der Pharmavertreter erzählte, dass die Angestellten angewiesen wurden „es nicht offensichtlich zu machen“, aber dass Geschenke und Gratisproben immer noch gang und gäbe seien.
Geschenke an ÄrztInnen und nicht-zertifizierte ÄrztInnen sind auf 2.000 indische Rupien (25 €) begrenzt. Es werden jedoch auch wesentlich kostspieligere Anreize geboten, etwa die Teilnahme an Fortbildungsprogrammen oder Zahlungen an Dritte. Manche ÄrztInnen erhalten bis zu 20.000 Rupien (250 €) Honorar, wenn sie auf Konferenzen sprechen.
Bei Sun Pharma wird von den VertreterInnen gefordert, jeden Tag bis zu einem Dutzend ÄrztInnen zu besuchen, dabei ist es egal ob diese qualifiziert sind oder nicht. Die Hälfte ihrer 200 regelmäßigen KundInnen sind informelle ÄrztInnen. Sie werden häufig noch energischer angegangen als richtige ÄrztInnen. Der Sun Pharma-Vertreter beschreibt nicht-zertifizierte ÄrztInnen als „einfachen“ Markt für Antibiotika und als eine Hauptzielgruppe. Professionelle ÄrztInnen müssten davon überzeugt werden, wie sicher und effektiv ein Medikament ist, während nicht-zertifizierte ÄrztInnen meistens keine Erklärungen verlangten, lediglich Anreize.
„Du kannst geben was du willst,“ berichtet er. „Von einer Nadel bis zu einer Rakete kannst du anbieten was du willst. Sie werden es annehmen. Und je mehr du reinsteckst, desto höher wird der Umsatz sein […]. Es kann ein Fernseher sei. Es kann ein Kühlschrank sein. Es kann alles Mögliche sein.“
Das aggressive Marketing der Pharma-Vertreter beruht auch auf den Verkaufszielen, die vom Unternehmen gesetzt werden: Sun Pharma VertreterInnen bekommen Boni wenn sie monatlich Antibiotika und Schmerzmittel im Wert von mehr als 300.000 Rupien (ca. 3.800 €) verkaufen.
Firmen verteidigen sich
Abbott distanziert sich offiziell von solchen Praktiken. Eine Sprecherin der Firma betonte, dass es den Vertriebsteams verboten sei, ÄrztInnen oder ApothekerInnen irgendetwas von Wert als Anreiz zu geben, um den Umsatz zu steigern. Außerdem erhielten die Teams regelmäßig Trainings zu Ethik und Compliance. VertriebsmitarbeiterInnen würden sich „nur mit lizensiertem Gesundheitspersonal treffen, die dazu autorisiert sind, Medikamente zu verschreiben“.
Sun Pharma erklärte, dass sie „überrascht und traurig waren über die dargelegten Ansichten eines angeblichen Vertreters von Sun Pharma“. Der Sprecher der Firma fügte hinzu: „Wir können kategorisch ausschließen, dass die Ansichten dieser Person in irgendeiner Weise den Werten von Sun Pharma entsprechen. Die angeblichen Aktivitäten stehen im Gegensatz zu unseren Unternehmensstatuten.“ Es sei verboten, Geschenke an das Gesundheitspersonal abzugeben, die „darauf zielen, unangemessen die Entscheidung zu beeinflussen, Produkte zu verschreiben, empfehlen, erwerben, bereitzustellen oder auszugeben“. Personal, die diese Regel oder ein Gesetz brächen, müssten mit Disziplinarmaßnahmen oder Entlassung rechnen.
Expertin bestätigt Probleme
Dr. Meenakshi Gautham, Forscherin an der London School of Hygiene and Tropical Medicine mit Sitz in Indien, hat über 10 Jahre lang nicht-registrierte ÄrztInnen interviewt. Sie kommt zu dem Schluss, dass Pharmafirmen es zunehmend auf diese Zielgruppe abgesehen haben und aggressiv Antibiotika vermarkten. „Pharma-Vertreter haben mir erzählt, wenn sie in manchen Gegenden das Ziel haben, Antibiotika im Wert von 100.000 Rupien (1.260 €) zu verkaufen, stammen 80.000 davon aus Verkäufen an informelle AnbieterInnen und 20.000 von registrierten ÄrztInnen.“
Gauthams Forschungsergebnisse zeigen, dass die meisten nicht-zertifizierten und privaten ÄrztInnen ihre gesamten Informationen über Antibiotika von PharmavertreterInnen und Pharmafirmen erhalten oder durch gesponserte Konferenzen. Ein Problem seien auch Gratisproben von Antibiotika, die die informellen ÄrztInnen erhalten. Sie geben die Proben dann häufig kostenlos an arme PatientInnen weiter. Doch die Proben reichen für gewöhnlich nicht für den kompletten Behandlungszyklus, die PatientInnen können damit also nicht ausreichend therapiert werden und der Resistenzbildung wird Vorschub geleistet.
Um die Probleme effektiv bekämpfen zu können, müssten solche aggressiven Vermarktungsstrategien der Firmen gestoppt werden. Erst dann kann es gelingen, in Zusammenarbeit mit den informellen Anbietern von Gesundheitsdienstleistungen den Antibiotika-Gebrauch zu reduzieren.
Über die AutorInnen:
Madlen Davies ist eine preisgekrönte Investigativjournalistin aus Großbritannien. Sie arbeitet für das Bureau of Investigative Journalism in Großbritannien. Einer ihrer Themenschwerpunkte sind die Gesundheitsrisiken durch resistente Erreger weltweit.
Ben Stockton ist britischer Investigativjournalist. 2019 gewann er gemeinsam mit Madlen Davies den Association of British Science Writers Preis. Beide wurden insbesondere für ihre Arbeit zu multiresistenten Keimen ausgezeichnet.
Rahul Meesaraganda arbeitet als unabhängiger Journalist in Andhra Pradesh, Indien. Seine Themenschwerpunkte sind Landwirtschaft, Umwelt und Wasser, mit einem Fokus auf die Situation in ländlichen Regionen. Auch zu Antibiotika-Resistenzen hat er bereits mehrere Berichte veröffentlicht, die internationale Aufmerksamkeit erlangten.
Madlen Davies & Rahul Meesaraganda sind PartnerInnen der BUKO Pharma-Kampagne im aktuellen Projekt zu Antibiotikaresistenzen.
Übersetzung: Hannah Eger
Bearbeitung: CJ/JS
Bilder © Madlen Davies
Artikel aus dem Pharma-Brief 10/2019, S. 1
[1] www.thebureauinvestigates.com
[2] www.thebureauinvestigates.com/stories/2019-08-19/drug-company-reps-give-quack-doctors-fridges-and-televisions-to-sell-antibiotics
[3] Laxminarayan R and Bhutta ZA (2016) Antimicrobial resistance - a threat to neonate survival. The Lancet Global Health; 4, p. e676
Länger prüfen bringt Erkenntnisse
Nachbeobachtungen von klinischen Studien
Studien zu therapeutischen Interventionen dauern oft nicht lange genug, um die Ergebnisse sicher interpretieren zu können. WissenschaftlerInnen aus Kanada und Australien haben jetzt untersucht, welche zusätzliche Erkenntnisse die Weiterverfolgung der TeilnehmerInnen nach Ende der Studie durch routinemäßig erhobene Patientendaten bietet.[1]
Wie lange klinische Studien dauern, ist oft von ökonomischen Interessen geprägt. Eine kürzere Dauer ist nicht nur preiswerter, vor allem kann sie zu einer früheren Zulassung führen. Das bedeutet mehr Geld für den Hersteller, denn eine längere Patentlaufzeit bedeutet ein längeres Monopol und damit höhere Einnahmen. Was bei der Eile nicht selten auf der Strecke bleibt, sind aussagekräftige Daten über den tatsächlichen Nutzen für die PatientInnen.
Die Nachverfolgung des Krankheitsverlaufs über das eigentliche Studienende hinaus bietet einen Realitätscheck. Werden die ersten Ergebnisse bei späteren Untersuchungen bestätigt oder widerlegt?
Voraussetzung für die Weiterverfolgung des Krankheitsverlaufs bei den Versuchspersonen nach Studienende sind krankheitsbezogene Register oder routinemäßig gespeicherte elektronische Patientenakten, die sich mit den Daten der klinischen Studie verknüpfen lassen. Entsprechend konzentrierte sich die fortgesetzte Beobachtung von PatientInnen auf wenige Länder, die solche Daten in größerem Umfang erfassen: Skandinavien, USA, Großbritannien, Niederlande, Australien, Neuseeland und Kanada.
Die AutorInnen fanden immerhin 113 Studien, bei denen eine anschließende systematische Nachverfolgung durchgeführt wurde. Die Bandbreite der geprüften Interventionen reichten von Medikamenten über Operationsverfahren bis zu Früherkennungsuntersuchungen. Auf der positiven Seite kann verbucht werden, dass in 28,4% der Studien ein signifikanter Nutzen erst bei der verlängerten Beobachtung festgestellt werden konnte. Andererseits löste sich bei 7,7% der Fälle der scheinbar vorhandene Nutzen in Nichts auf. Und in 9% der Fälle wurden unerwünschte Wirkungen erst nachträglich erkannt. Bei 21,9% bestätigte sich die bereits bei Studienende prognostizierte Nutzlosigkeit bzw. der fehlende Vorteil der Intervention.
Auch wenn in den meisten Nachbeobachtungen die Randomisierung (Zuordnung der PatientInnen zu den Studienarmen nach dem Zufallsprinzip) aufrecht erhalten wurde, bleiben Einschränkungen: Die Verblindung war nicht immer gewährleistet, und die weitere Behandlung der PatientInnen kann unterschiedlich gewesen sein. Auch die Vollständigkeit der Registerdaten ist fraglich, allerdings werden schwerere Ereignisse wie Krebserkrankungen, Herzinfarkte oder Schlaganfälle eher erfasst.
In vielen Fällen war unklar, ob nur bestimmte handverlesene Ergebnisse weiter beobachtet wurden, Rosinenpickerei ist also nicht ausgeschlossen. Trotz allem birgt diese Analyse eine wichtige Botschaft: Man kann den ersten Ergebnissen von Studien nicht immer vertrauen. (JS)
Artikel aus dem Pharma-Brief 2/2019, S. 7
[1] Fitzpatrick T et al. (2018) Assessment of long-term follow-up of randomized trial participants by linkage to routinely collected data. JAMA open; 1, p e186019, doi: 10.1001/jamanetworkopen.2018.6019
Klinische Studien:
Universitäten lassen Transparenz vermissen
Zum Schutz von PatientInnen müssen die Ergebnisse aller klinischen Studien veröffentlicht werden. Doch eine Untersuchung von europäischen Universitäten zeigt, dass viele diese Verpflichtung schlecht erfüllen. Ein gemeinsamer Bericht von BUKO Pharma-Kampagne, TranspariMED (Vereinigtes Königreich), Test Aankoop (Belgien) und Health Action International (Amsterdam) macht gravierende Defizite deutlich. Untersucht wurden die 30 Universitäten, die die meisten Studien durchführen.[1]
Klinische Studienergebnisse nicht zu melden, hat erhebliche negative Konsequenzen für PatientInnen und die öffentliche Gesundheit. Seit Juli 2014 schreibt die Europäische Union (EU) vor, dass für jede klinische Studie, die im EU Clinical Trials Register eingetragen ist, eine Zusammenfassung ihrer Ergebnisse innerhalb von 12 Monaten nach Studienabschluss (sechs Monate für pädiatrische Studien) im europäischen Studienregister veröffentlichen muss. Diese Regulierung trifft auch auf Studien zu, die vor 2014 abgeschlossen wurden und gilt unabhängig davon, ob die Studienergebnisse bereits in der Fachliteratur publiziert wurden. Das bedeutet, dass klinischen Studien, bei denen die Sponsoren keine Zusammenfassung der Ergebnisse im EU-Register veröffentlichten, gegen die Transparenzverordnung der EU verstoßen.
Die Ergebnisse im Überblick
Von den 30 europäischen Universitäten, die die meisten Studien gemeldet haben, fehlen insgesamt nachweisbar Ergebnisse von 778 fälligen klinischen Studien im europäischen Studienregister, das entspricht 83% (Tabelle 1). Lässt man die Universitäten im Vereinigten Königreich außen vor, liegt die Melderate bei lediglich 7%. Die absolute Zahl an fehlenden Ergebnissen von fälligen Studien ist vermutlich deutlich höher.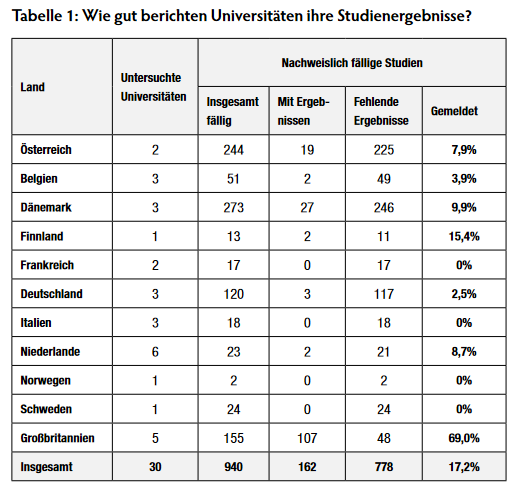
Nur drei Universitäten schneiden gut ab: University of Oxford, University College London und King’s College London. Diese Einrichtungen haben bereits über 80% ihrer fälligen Studienergebnisse veröffentlicht. 14 der 30 untersuchten Universitäten haben dagegen nicht ein einziges Studienergebnis veröffentlicht. Das trifft auf alle Universitäten in Frankreich, Italien, Norwegen und Schweden zu. Auch die verbliebenen 13 Universitäten schneiden mit Melderaten von 2 – 33% schlecht ab.
Die Tatsache, dass britische Universitäten ihre europäischen Peers mit großem Abstand übertreffen, liegt am Druck des Parlaments, der Forschungsförderer und der Öffentlichkeit. Die starke Performance einiger Universitäten im Vereinigten Königreich zeigt, dass Universitäten in ganz Europa es deutlich besser machen können – und auch müssen.
Empfehlungen
Universitäten sollten die Ergebnisse von allen klinischen Studien – vergangene, gegenwärtige und zukünftige – in allen Registern veröffentlichen, in denen die Studien eingetragen wurden. Bei laufenden und künftigen Studien sollten Universitäten die Ergebnisse innerhalb von zwölf Monaten nach Abschluss veröffentlichen.
Darüber hinaus sollten die Universitäten die gemeinsame Erklärung der International Clinical Trials Registry Platform (ICTRP) der Weltgesundheitsorganisation (WHO)[2] unterzeichnen und die dort beschriebenen Regeln zu Transparenz befolgen.
Nationale Zulassungsbehörden sollten regelmäßig den Status aller Studien überprüfen, die in ihren Registern noch als „laufend“ gelistet sind, und ihren Status – falls erforderlich – in „abgeschlossen“ ändern.
Nationale Forschungsförderer sollten ebenfalls die gemeinsame Erklärung der WHO unterzeichnen, um PatientInnen zu schützen und zu verhindern, dass von SteuerzahlerInnen finanzierte medizinische Forschung wegen Nichtveröffentlichung der Ergebnisse der Studien verschwendet werden.
Nationale Regierungen sollten systematisch kontrollieren, ob klinische Studien, die in ihrem Zuständigkeitsbereich durchgeführt werden, ihre Studienergebnisse in öffentlichen Registern innerhalb von zwölf Monaten veröffentlichen, wie es die Best Practices der WHO vorsehen. Studiensponsoren, die ihre Ergebnisse nicht innerhalb der Frist veröffentlichen, sollten sanktioniert werden. Das Vereinigte Königreich bereitet entsprechende Schritte gegenwärtig vor.
Wie gut sind die Länder?
Dieser Bericht untersucht das Meldeverhalten von den 30 europäischen Universitäten mit den klinischen Studien, die der EU-Verordnung zu klinischen Studien unterliegen. Insgesamt haben diese Universitäten 4.575 klinische Studien gesponsert. Für 940 dieser Studien sind Ergebnisse nachweislich fällig. Allerdings haben lediglich 162 der nachweisbar fälligen Studien (17%) ihre Ergebnisse im EU Clinical Trials Register veröffentlicht. Bei den verbleibenden 778 Studien (83%) verstoßen die Universitäten somit gegen die Transparenzvorschriften der EU.
Die meisten der 778 Studien mit nachweislich fehlender Ergebnisberichterstattung wurden von Universitäten in Dänemark (246 Studien), Österreich (225) und Deutschland (117) durchgeführt. Von den untersuchten Universitäten in Frankreich, Italien, Norwegen und Schweden wurde kein einziges Studienergebnis im Register veröffentlicht. Manche Universitäten in Belgien, Deutschland und den Niederlanden haben ebenfalls für keine ihrer klinischen Studien Ergebnisse veröffentlicht. Nur ein paar britische Universitäten haben gut abgeschnitten, manche Institutionen rühmen sich mittlerweile mit Melderaten über 80%. Schließt man die britischen Universitäten von der Betrachtung aus, liegt die durchschnittliche Melderate in Europa gerade mal bei 7% und somit sogar niedriger als die 11%, die Forscher für europäische Universitäten im September 2018 ermittelt haben. Außerhalb des Vereinigten Königreichs fehlen Ergebnissen zu 730 von 785 nachweisbar fälligen Studien (93%).
In Deutschland
Von den einbezogenen drei deutschen Universitäten schneidet München mit 7% noch am besten ab, die Charité kommt auf 2% und Heidelberg hat kein einziges Ergebnis veröffentlicht. Positiv sticht in Deutschland nur die Uni Münster ab, die nicht unter den Top 30 Forschungsstätten ist (und deshalb in die vorliegende Untersuchung nicht mit einbezogen wurde). Sie hat 58,8% aller Studien veröffentlicht.[3]
Die obenstehenden Zahlen unterschätzen vermutlich signifikant den wahren Anteil an klinischen Studien, die wegen fehlender Veröffentlichung der Ergebnisse gegen die EU Regeln verstoßen. Das liegt daran, dass zahlreiche Studien als „laufend“ im europäischen Studienregister gelistet werden, obwohl sie schon längst abgeschlossen sind. Zum Beispiel haben Universitäten in den Niederlanden insgesamt 967 Studien durchgeführt, aber nur 23 davon (2,4%) sind als „abgeschlossen“ gekennzeichnet. Dieser geringe Anteil ist nicht plausibel, da die Registereinträge zeigen, dass viele dieser Studien vor über fünf Jahren begonnen wurden. Im Vereinigten Königreich, wo das nationale Register gerade aktualisiert wird, liegt der Anteil der „abgeschlossenen“ Studien bei 27,4%.
Im aktuellen Meldesystem laden die Universitäten direkt ihre zusammengefassten Ergebnisse in das EU Register hoch – als Studiensponsoren sind sie rechtlich dazu verpflichtet und der Prozess liegt komplett in ihrer eigenen Kontrolle. Allerdings können Universitäten nicht direkt den Status ihrer Studien (laufend, abgeschlossen) aktualisieren. Stattdessen müssen sie ihre nationale Zulassungsbehörde darüber informieren, dass eine Studie abgeschlossen ist. Die Behörde aktualisiert dann den Status der Studie im Register. In Ländern mit unerklärlich niedrigen Anteilen an „abgeschlossenen“ Studien, wie die Niederlande, ist es sehr wahrscheinlich, dass die nationalen Behörden eine große Zahl von Registereinträgen nicht geändert haben, obwohl die Studien abgeschlossen waren. Diese Behörden sollten dem positiven Beispiel der britischen MHRA folgen, und systematisch den Status aller klinischen Studien, die in ihrem Land durchgeführt wurden, durchsehen und aktualisieren.
Warum sind britische Universitäten besser?
Im Durchschnitt schneiden die britischen Universitäten deutlich besser ab als die in anderen Ländern. Die schwächste der fünf untersuchten Universitäten im Vereinigten Königreich hat eine Melderate von nur 25%, die zwei Stärksten – University of Oxford und King’s College London – haben bisher schon über 90% der fälligen Studienergebnisse veröffentlicht. Zum Vergleich: von einer Ausnahme abgesehen, hat nicht eine einzige Universität des europäischen Festlands eine höhere Melderate als 20%.
Britische Universitäten haben in Sachen Transparenz eine Führungsposition in Europa eingenommen. Ursache dafür war der Druck des Parlaments, der Forschungsförderer und der Öffentlichkeit.
Druck vom Parlament
Das Science and Technology Committee des britischen Parlaments hat 2018 -2019 eine Untersuchung zu wissenschaftlicher Integrität durchgeführt. Mitglieder des Ausschusses waren schockiert, als sie herausfanden, dass zahlreiche Universitäten regelmäßig gegen die Transparenzregeln verstoßen. Anfang 2019 warnte der Vorsitzende des Ausschusses die britischen Universitäten mit einem Schreiben, dass sie vor den Ausschuss zitiert würden und sich rechtfertigen müssten, wenn sie die fehlenden Studienergebnisse nicht bis Sommer 2019 in das Register einstellten.[4]
Druck von den Forschungsförderern
Die zwei öffentlichen medizinischen Forschungsförderungsorgane im Vereinigten Königreich, das NIHR und der MRC, sowie der private Wellcome Trust haben 2017 die gemeinsame Erklärung der WHO zur Veröffentlichung von Studienergebnissen2 unterzeichnet. Mit ihrer Unterschrift verpflichten sich die Forschungsförderer, die Regeln zur Studienregistrierung und Ergebnismeldung gemäß der WHO Best Practices zu befolgen und zu kontrollieren, ob die geförderten Projekte diese Regeln befolgen. Der MRC hat bereits ein exzellentes Review zu den von ihm geförderten klinischen Studien durchgeführt.[5] In den folgenden Jahren könnten britische Universitäten, die es versäumen ihre Studienergebnisse rechtzeitig in den Registern zu veröffentlichen, von weiteren Förderungen ausgeschlossen werden.
Öffentlicher Druck
Eine von TranspariMED initiierter loser Zusammenschluss von Gruppen, die sich um Integrität in der Gesundheitsversorgung kümmern, darunter Universities Allied for Essential Medicines (UAEM-UK), HealthWatch UK, Transparency International Health und STOPAIDS, hat sich an das Parlament gewendet, die Medien informiert und Druck auf die Universitäten ausgeübt, um eine bessere Meldepraxis für Studienergebnisse zu erreichen.
TranspariMED und UAEM-UK haben zudem mehrere Berichte zur Leistung einzelner britischer Universitäten veröffentlicht.[6] Zeitgleich hat sich die hauptsächlich in Großbritannien aktive AllTrials Kampagne[7] des Problems angenommen, unter anderem mit regelmäßigen E-Mails an ihre über 90.000 Unterstützer. Das EBM Data Lab der University of Oxford hat den EU Trials Tracker konzipiert, von dem die Daten für den vorliegenden Bericht stammen. Das EBM Data Lab arbeitet mit der AllTrials Kampagne zusammen und hat dem Parlamentsausschuss mit den notwendigen Daten zur Veröffentlichungspraxis von klinischen Studien der Universitäten versorgt.
Der gebündelte Druck hatte einen enormen Einfluss auf das Meldeverhalten der britischen Universitäten. Das King’s College London beispielsweise hat innerhalb nur eines halben Jahres seine Melderaten von 18% auf 93% verbessert. Die University of Nottingham, die vom Parlamentsausschuss 2018 wegen ihrer schlechten Leistung angeprangert wurde, hat inzwischen die Ergebniszusammenfassungen zu über 95% der von der Uni durchgeführten Studien veröffentlicht.
Soweit TranspariMED das beurteilen kann, arbeitet momentan jede medizinische Universität im Vereinigten Königreich hart daran, fehlende Studienergebnisse im EU Register hochzuladen und oftmals sogar in anderen Registern, wie das ISRCTN und das US Register Clinicaltrials.gov. Dadurch wird deutlich, dass wo ein Wille ist, auch ein Weg ist – andere Universitäten in Europa können also ebenfalls die Probleme lösen, wenn sie wollen. (Siehe Artikel zur Nottingham University auf S. 10.)
Momentan arbeitet die britische Regierung daran, ein umfassendes nationales Monitoringsystem für klinische Studien einzuführen. Es soll jede einzelne klinische Studie, die auf britischem Boden durchgeführt wird, nachverfolgen, einschließlich kommerzieller und länderübergreifender Studien. Damit soll sichergestellt werden, dass alle Studien registriert und ihre Ergebnisse veröffentlicht werden.
Relevanz für die Praxis
Da Verschweigen von Studienergebnissen ist kein Kavaliersdelikt. Ein Bericht von 2017 von Transparency International und Cochrane zeigt, dass es fehlende Ergebnisberichte von Studien gravierende negative Konsequenzen haben können:[8] Patienten geraten durch falsche Behandlungsentscheidungen in Gefahr, öffentliche Einrichtungen können keine informierten Entscheidungen über den Nutzen von Arzneimitteln treffen, öffentliche Gelder werden verschwendet und der medizinische Fortschritt wird verlangsamt.
Rechtliche Rahmenbedingungen
Eine EU-Verordnung schreibt seit Juli 2014 vor, dass eine Zusammenfassung der Ergebnisse für jede einzelne Studie, die im EU Studienregister EudraCT eingetragen ist, innerhalb von zwölf Monaten (bei pädiatrischen Studien sechs Monate) nach Studienabschluss veröffentlicht werden muss. Das bedeutet, dass alle in diesem Bericht ermittelten klinischen Studien, bei denen eine Zusammenfassung der Ergebnisse fehlt, gegen die Transparenzverordnung der EU verstoßen. Sie wurde eingeführt, um die Interessen der PatientInnen und SteuerzahlerInnen zu schützen.
Verschwendete Forschung
Ungemeldete Studien hemmen den Fortschritt in der Wissenschaft, schaden der öffentlichen Gesundheit und sind daher eine Verschwendung von Forschungsgeldern. In der Vergangenheit haben das Verschweigen der Ergebnisse von klinischen Studien zum Tod unzähliger PatientInnen und Verluste im Wert von Milliarden Euros verursacht.[9] Aus diesem Grund hat die Erklärung von Helsinki das Melden von Ergebnissen jeder klinischen Studie zu einer universeller ethischen Pflicht für alle medizinischen ForscherInnen weltweit erklärt.[10]
Nicht alle Studien, die ihre Ergebnisse nicht im EU Studienregister eingetragen haben, sind komplett unveröffentlicht. Jedoch weist die bestverfügbare Evidenz darauf hin, dass etwa von der Hälfte aller Studien, deren Ergebnisse im EU Register fehlen, auch nicht in Fachzeitschriften veröffentlicht wurden. Dadurch geraten Hunderte von Studien an europäischen Universitäten in Gefahr, eine Verschwendung von Forschungsgeldern zu werden, werden ihre Ergebnisse nicht bald veröffentlicht.
Universitäten müssen dringend ihre klinischen Studien im EU-Register, dem US-Register clinicaltrials.gov und anderen WHO Primärstudienregistern durchsehen, ungemeldete Studien identifizieren und sicherstellen, dass fehlende Eintragungen nachgeholt und die Ergebnisse einer Studie so bald wie möglich nach deren Abschluss veröffentlicht werden.
Global Best Practices
Die WHO Standards schreiben vor, dass die Ergebnisse jeder Interventionsstudie in jedem öffentlichem Register, in dem sie eingetragen sind, zwölf Monate nach Abschluss der Studie veröffentlicht werden. Die WHO hebt hervor, dass die Veröffentlichung von Studienergebnissen in der Fachliteratur kein Ersatz zur Meldung der Ergebnisse in öffentlichen Registern darstellt.
Best Practices, die gemeinsam von Cochrane und Transparency International entwickelt wurden, sehen ebenfalls vor, dass „eine Zusammenfassung der Ergebnisse für alle klinischen Studien in den Registern, in denen sie ursprünglich registriert wurden, innerhalb von zwölf Monaten nach Studienabschluss hochgeladen werden muss.“8 Beide Organisationen machen darauf aufmerksam, dass das nachträgliche Hochladen der Ergebnisse von allen vergangenen Studien in den Registern „die Gesundheitsversorgung und die Entscheidungsfindung von Regierungseinrichtungen zur Ressourcenverteilung verbessern würde, und gleichzeitig würden Milliarden von Dollar für medizinische Forschung davor bewahrt, für immer verloren zu sein.“
Übereinstimmend damit fordert die AllTrials Kampagne, dass „eine Zusammenfassung der Ergebnisse […] dort wo die Studie registriert wurde innerhalb eines Jahres nach Studienabschluss hochgeladen werden sollte.“
Es gibt gute Gründe dafür, dass alle Studienergebnisse in den Registern veröffentlicht werden sollten. Das Hochladen der Ergebnisse in den Registern beschleunigt den medizinischen Fortschritt, denn durch die 12-Monatsfrist können Ergebnisse deutlich schneller geteilt werden. Der Veröffentlichungsprozess in der Fachliteratur ist viel langwieriger.
Das Risiko, dass die Ergebnisse einer Studie nie veröffentlicht werden und eine Verschwendung von Forschung darstellen, wird minimiert. Das kann sonst zum Beispiel passieren, wenn der Studienleiter während des langwierigen Prozesses einer wissenschaftlichen Veröffentlichung stirbt oder seine Stelle wechselt.
Forschung hat ergeben, dass Studienergebnisse, die in Register hochgeladen wurden, üblicherweise ein verständlicheres und genaueres Bild der Ergebnisse, die für PatientInnen relevant sind, geben als wissenschaftliche Publikationen. Studienergebnisse in den Registern sind einfacher aufzufinden und öffentlich zugänglich. Das Melden in Registern vereinfacht es, die Studienergebnisse mit den ursprünglich festgelegten Zielen zu vergleichen. Somit werden schädliche Verletzungen der Sorgfaltspflicht in der Forschung, wie das „stille“ Zurückhalten unvorteilhafter Ergebnisse, Hinzufügen oder Ändern der ursprünglich festgelegten Endpunkte,[11] selektives Berichten von für den Sponsor günstigen Ergebnissen und Praktiken wie HARKing[12] und p-hacking,[13] weniger wahrscheinlich.[14]
Falsche Vorwände
Das International Committee of Medical Journal Editors befürwortet das Hochladen von Studienergebnissen in Studienregistern und betont, dass das nicht als Vorveröffentlichung eines Artikels betrachtet wird. Das bedeutet, dass wissenschaftliche Zeitschriften Artikel akzeptieren, auch wenn die Ergebnisse zuvor im Studienregister veröffentlicht wurden.[15] Da das Melden der Ergebnisse in den Registern üblicherweise schneller ist als eine wissenschaftliche Publikation, ist es nun Best Practice für wissenschaftliche Kommunikation, die Studienergebnisse in Registern zu veröffentlichen, bevor sie in der Fachliteratur erscheinen.
Der vorliegende Text wurde ursprünglich von Till Bruckner verfasst, von Hannah Eger übersetzt und von Jörg Schaaber überarbeitet. c Er unterliegt wie das Original [16] einer Creative Commons BY 3.0 Lizenz.
Artikel aus dem Pharma-Brief 4-5/2019, S.5
Bild window cleaning © Vikramjit Kakati
[1] Die Untersuchung wurde von Till Bruckner (TranspariMED) durchgeführt. Der vorliegende Text ist eine überarbeitete und leicht gekürzte Fassung des englischen Originals.
[2] WHO (2017) International Clinical Trials Registry Platform (ICTRP) Joint statement on public disclosure of results from clinical trials www.who.int/ictrp/results/jointstatement/en
[3] http://eu.trialstracker.net/sponsor/university-of-munster
[4] Bruckner T (2019) UK government promises national strategy to boost clinical trial reporting. 25 Feb. www.transparimed.org/single-post/2019/02/25/UK-government-promises-national-strategy-to-boost-clinical-trial-reporting
[5] MRC (2017) MRC Review of Clinical Trials. https://mrc.ukri.org/research/policies-and-guidance-for-researchers/review-of-clinical-trials
[6] www.transparimed.org/resources
[8] TI (2017) Clinical trials transparency https://docs.wixstatic.com/ugd/01f35d_def0082121a648529220e1d56df4b50a.pdf
[9] Bruckner T and Ellis B (2017) Clinical Trial Transparency https://media.wix.com/ugd/01f35d_0f2955eb88e34c02b82d886c528efeb4.pdf
[10] WMA (2019) Declaration of Helsinki www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects
[11] Altman DG et al. (2017) Harms of outcome switching in reports of randomised trials: CONSORT perspective. BMJ;356, p j396
[12] HARKing = Hypothesizing after the results are known. Neue Hypothesen aufstellen, wenn man die Ergebnisse schon kennt
[13] p-hacking. So lange mit den Daten spielen, bis das statistische Signifkanzniveau (p-Wert) doch noch erreicht wird. Üblicherweise gilt ein p-Wert unter 0,05 als signifikant (das Ergebnis ist dann zu 95% nicht zufällig)
[14] Munafò MR et al. (2017) A manifesto for reproducible science. Nature Human Behaviour; 1, article number 0021 www.nature.com/articles/s41562-016-0021
[15] ICMJE (2019) Clinical Trials www.icmje.org/recommendations/browse/publishing-and-editorial-issues/clinical-trial-registration.html [Zugriff 24.7.2019]
[16] www.bukopharma.de/images/pressemitteilungen/2019/Clinical_Trial_Transparency_EU_Universities_2019.pdf
Kaum Regeln zum Umgang mit Interessenkonflikten
Big Pharma beeinflusst Medizinstudierende – Universitäten schauen weg
Pharmafirmen üben durch Werbung und Sponsoring bereits während des Medizinstudiums Einfluss auf die ÄrztInnen von morgen aus. Eine neue Studie der Studierendenorganisation Universities Allied for Essential Medicines (UAEM) zeigt, dass medizinische Fakultäten in Deutschland kaum etwas unternehmen, um ihre Studierenden vor kommerzieller Beeinflussung zu schützen.
Eine aktuelle Publikation von UAEM macht die Versäumnisse deutscher Universitäten deutlich: Die Studierendenorganisation untersuchte den Umgang mit Interessenkonflikten an 38 medizinischen Fakultäten und bewertete deren institutionalisierte Regeln zum Umgang mit Interessenkonflikten anhand eines Punktesystems. Zur Ermittlung der Daten kontaktierten die AutorInnen die Dekanate der Fakultäten und recherchierten zusätzlich auf deren Webseiten anhand einschlägiger Suchbegriffe. Das Resultat: Lediglich 16 der 38 angefragten Universitäten gaben überhaupt eine Rückmeldung, ob Regelungen zu Interessenkonflikten vorhanden seien. Davon erfüllte jedoch nur eine Regelung der Technischen Universität Dresden die gesetzten Einschlusskriterien. Bei der zusätzlichen webbasierten Suche erfüllte nur die Charité-Universitätsmedizin Berlin die Einschlusskriterien der Studie.
Gerade einmal zwei der 38 medizinischen Fakultäten in Deutschland verfügen demnach über Statuten, anhand derer sich die Studierenden in dem Umgang mit der Pharmaindustrie orientieren können. Doch auch diese Fakultäten kommen bei der qualitativen Bewertung anhand des Punktesystem nicht gut weg: Die TU Dresden erhielt 12 von 26 Punkten, die Charité Berlin 4 von 26.[1]
Firmen machen schon Studierenden Avancen
Dabei wäre es dringend geboten, bereits angehende MedizinerInnen im Umgang mit Interessenkonflikten zu schulen und gegen die Avancen der Industrie entsprechend zu rüsten. Denn die Hersteller versuchen die zukünftigen ÄrztInnen schon früh abzuholen. In einer Umfrage an acht medizinischen Fakultäten in Deutschland fanden Lieb und Koch schon vor mehreren Jahren heraus, dass lediglich 12% der Befragten nie ein Geschenk von der Pharmaindustrie erhalten oder an einer gesponserten Veranstaltung teilgenommen hatten.[2] Einer Studie von Jahnke et al. zufolge, waren 77% der Medizinstudierenden vor Ende ihrer klinischen Ausbildung mindestens einmal mit VerkaufsvertreterInnen eines Pharmakonzerns konfrontiert gewesen. 13% waren sogar außerhalb der Universität direkt kontaktiert worden.[3]
Das Medizinstudium trägt jedoch wenig dazu bei, angehende MedizinerInnen vor solcher Beeinflussung zu schützen: 65% der Studierenden fühlen sich nicht gut auf den Umgang mit der pharmazeutischen Industrie vorbereitet und 90% berichten, dass der Umgang mit PharmavertreterInnen und deren Werbepraktiken nie in einer Vorlesung thematisiert wurde.[3]
Interessenkonflikte kommen in der Lehre nicht vor
Es ist also schon seit mehreren Jahren bekannt, dass hier großer Bedarf besteht. Schließlich sind Interessenkonflikte gerade im Gesundheitswesen besonders häufig. Denn das Interesse der PatientInnenpflege kollidiert nicht selten mit dem Bestreben der Industrie, Arzneimittel oder Medizinprodukte gewinnbringend zu verkaufen. Gerade ÄrztInnen sind mit massivem Marketing der Hersteller konfrontiert – die Bandbreite der Beeinflussungsstrategien reicht von kleinen Geschenken, die das Verschreiben bestimmter Arzneimittel versüßen über das Sponsoring von Fortbildungen und fürstlich entlohnten Anwendungsbeobachtungen bis hin zur Praxis-Software, die mit Werbeanzeigen bespickt ist.[4] Der kritische Blick auf solche Werbestrategien tut Not. Denn nur, wer früh lernt, Beeinflussung zu durchschauen und deren Folgen kennt, kann sich auch wirksam davor schützen. (MB/CJ)
Artikel aus dem Pharma-Brief 7-8/2019, S.2
[1] Grabitz P et al. (2019) Conflict of Interest Policies at German medical schools – A long way to go. www.biorxiv.org/content/10.1101/809723v1 [Zugriff 22.10.2019]
[2] Lieb K and Koch C (2013) Medical Students’ Attitudes to and Contact With the Pharmaceutical Industry. Deutsches Ärzteblatt International; 110, p 586 http://dx.doi.org/10.3238/arztebl.2013.0584
[3] Jahnke K et al. (2014) Exposition und Einstellungen deutscher Medizinstudierender zu Pharmawerbeaktivitäten: Eine Querschnittsstudie. GMS Zeitschrift für Medizinische Ausbildung; 31, S. 10 http://dx.doi.org/10.3205/zma000924
[4] Berndt C und Grill M (2019) Medizinische Fakultäten ignorieren Interessenkonflikt. Süddeutsche Zeitung. www.sueddeutsche.de/wissen/pharmaindustrie-universitaeten-interessenkonflikte-1.4644884 [Zugriff 22.10.2019]
Interessenkonflikte und der Schutz der öffentlichen Gesundheit
Interview mit Judith Richter
Judith Richter arbeitet seit vielen Jahren zum Thema Interessenkonflikte mit Schwerpunkt auf internationale Organisationen. Wir haben sie befragt, wie sich die Situation in den letzten Jahrzehnten verändert hat, welche negativen Folgen das hat und was zu einer Verbesserung der Lage notwendig wäre.
Ich habe gehört, dass du in den letzten Jahren, versucht hast, das Konzept der Interessenkonflikte auf zahlreichen Konferenzen und in Schulungen zu erklären. Wie und wann begann dein Interesse an diesem Thema?
Als Person, die im Gesundheitsbereich arbeitet, bin ich schon lange an Interessenkonflikten interessiert gewesen, auch wenn ich damals diesen Begriff noch nicht benutzte. Als ich zum Beispiel in den siebziger Jahren Vizepräsidentin des Schweizerischen Pharmaziestudentenverbandes (ASEP) war, verfochten wir die Ansicht, dass Apotheker es ablehnen sollten, ihre Schaufenster von Pharmafirmen dekorieren zu lassen. Wir befürchteten, die KundInnen könnten Apotheken als Outlet von Pharmafirmen ansehen und nicht als Orte, die eine unabhängige Gesundheitsberatung anbieten.
In den 1990er Jahren bat mich IBFAN-Europe (International Baby Food Action Network)[1] zu seinen internen Diskussionen über Interessenkonflikte beizutragen: Wie könnten IBFAN-Mitglieder zum Beispiel am besten thematisieren, dass die Hersteller von Muttermilchersatzprodukten sowohl Kinderarztverbände als auch deren Weiterbildungen sponserten? Wir diskutierten auch die möglichen Interessenkonflikte durch finanzielle Verbindungen von StillberaterInnen mit Herstellern von Milchpumpen.
Damals wurde ich auch von Health Action International Europe eingeladen, meine Bedenken bezüglich des Sponsorings durch Pharmaunternehmen und reiche Unternehmensphilanthropen wie Bill Gates darzulegen.[2]
Das Thema Interessenkonflikte wurde also schon damals breit diskutiert?
In der Tat. Und etliche ForscherInnen im Bereich Interessenkonflikte (Conflict of Interest, CoI) betonen, dass der Aufstieg des neoliberalen Wirtschaftsmodells die Anzahl bereits bestehender Interessenkonflikte noch vergrößert und neue Typen geschaffen hat.
Meine Arbeit als Soziologin zu adäquatem und wirksamem Schutz öffentlicher Interessen – einschließlich der Regulierung von Interessenkonflikten – begann 2002. Damals hatte ich gerade eine längere Beratertätigkeit für UNICEF über die Möglichkeiten effektiver Regulierung der Marketingmethoden transnationaler Säuglings- und Kindernahrungskonzerne beendet.[3] Durch diese Arbeit wurde ich auf die immer engeren Verbindungen von UN-Organisationen mit transnationalen Firmen aufmerksam. Sie riskierten dadurch ihr eigentliches Mandat zu vernachlässigten, die Aktivitäten eben dieser Konzerne durch internationale Regulierungen zu kontrollieren.
Mein Anliegen – und auch zum Beispiel das der finnischen Forscherin Eeva Ollila[4] – war damals, auf die vielfältigen Risiken aufmerksam zu machen, die sogenannte Globale Öffentlich-Private Partnerschaften für Gesundheit (GHPPP) und Multi-Stakeholder Initiativen (MSIs) mit sich bringen: sie untergraben die Fähigkeit öffentlicher Institutionen, im öffentlichen Interesse zu handeln.
Die finnische Regierung bat mich, zu untersuchen, welche Möglichkeiten es gebe, dies abzuwenden. Ich fand heraus, dass die damalige Generaldirektorin der WHO, Dr. Brundtland, den WHO Mitgliedsstaaten versichert hatte, die WHO werde Richtlinien und Trainingskurse entwickeln, um Interessenkonflikte zu erkennen und zu vermeiden. Als ich jedoch Mitarbeiter der WHO dazu interviewte, teilten sie mir mit, dass dieses Projekt gestoppt worden war. Ihnen wurde damals eingeimpft, sie sollten transnationale Firmen im Gesundheitsbereich lieber als ‘Partner’ ansehen und nicht als profitorientierte Akteure. Das Achten auf Interessenkonflikte wurde in diesem Modell als Hindernis gesehen, auf ‚flexiblere Art und Weise‘ zu arbeiten. Ein leitender juristischer Funktionär der WHO teilte mir mit, dass die existierenden CoI Definitionen als ‘zu einschränkend’ wahrgenommen würden. Mitarbeiter auf unterer Ebene befürchteten dagegen, dass sie nicht in der Lage sein würden, problematische Vorschläge von Großunternehmen und reichen Sponsoren abzuwehren.[5]
Das motivierte mich, existierende Interessenkonflikts-Theorien und Richtlinien zu untersuchen. Ich hoffte, das könnte helfen, eventuelle Missverständnisse zu klären, die die WHO davon abhielten, ihre Beziehungen mit Firmen und Unternehmensphilanthropen angemessen zu regulieren.
Etwas später bat mich das Genfer Regionalbüro von IBFAN, zum Paragraph 35 der WHO/UNICEF Globalen Strategie zur Ernährung von Säuglingen und Kleinkindern zu arbeiten.[6] Dieser Artikel besagt, dass „alle Partner zusammenarbeiten sollten“ – auch indem sie Allianzen und „Partnerschaften“ formen – „im Einklang mit akzeptierten Prinzipien zur Vermeidung von Interessenkonflikten.“ Jedoch waren diese Prinzipien nirgendwo präzisiert. Ich untersuchte, wie man hier Abhilfe schaffen könnte und veröffentlichte “Conflicts of Interest and Policy Implementation – reflections from the fields of health and infant feeding“.[7]
Kannst du eine brauchbare Definition von Interessenkonflikten geben?
In oben genannter Publikation wies ich im Wesentlichen auf drei Definitionen hin: Eine wurde entwickelt für den öffentlichen Dienst in den Ländern der Organisation für Wirtschaftliche Zusammenarbeit und Entwicklung[8] (OECD) und zwei Definitionen stammen aus dem Bereich der Gesundheit.
Die erste Definition, wurde entwickelt von dem politischen Philosophen Professor Dennis F. Thompson.[9] Ich fand seine Definition von Interessenkonflikten als Konflikten zwischen primären und sekundären Interessen nützlich, um zu erklären, warum man über Konflikte von „Interessen“ spricht. Diese Definition hat sich mit geringfügigen Änderungen seitdem weit verbreitet durch den Bericht des Kommitees des US Institute of Medicine (IoM) zu Interessenkonflikten in der medizinischen Forschung, Ausbildung und Praxis.[10] „Interessenkonflikte sind definiert als Gegebenheiten, die ein Risiko dafür schaffen, dass professionelles Urteilsvermögen oder Handeln, welches sich auf ein primäres Interesse beziehen, durch ein sekundäres Interesse unangemessen beieinflusst wird“ [11]
Wie du weisst, haben kritische deutsche ÄrztInnen die Thompson-IoM Definition in ihrer Arbeit zur besseren Identifizierung von Interessenkonflikten im medizinischen Bereich übernommen und viele nützliche Ratschläge weiterentwickelt. [11]
Die andere Definition, die ich damals fand, wurde von dem Rechtsprofessor Marc Rodwin formuliert. Sein Buch „Medicines, Money & Morals: Physicians‘ Conflicts of Interest“ [12] wurde 1993 veröffentlicht, im gleichen Jahr wie der vielzitierte Artikel von Thompson. Rodwin fasste seine Konzeption kürzlich nochmals wie folgt zusammen: „Ein Interessenkonflikt existiert, wo ein Individuum eine Verpflichtung hat, einer Partei zu dienen oder eine Rolle zu spielen, und das Individuum andererseits: 1) Anreize oder 2) widersprüchliche Loyalitäten hat, die das Individuum ermutigen, in einer Art und Weise zu handeln, mit der es seine Verpflichtungen bricht.“ [13]
Wie mir Professor Rodwin erklärte, folge er damit der Begriffsauffassung, die im amerikanischen Recht seit dem 20. Jahrhundert üblich ist. Diese rechtliche Definition ist etwas komplexer, das stimmt. Das US Institute of Medicine hatte die Thompson-IoM Definition als die „zweckmäßigste“ hervorgehoben. Ich hatte nie die Gelegenheit zu fragen, warum die IoM Komission anscheinend die Konzeption von Professor Rodwin nicht in ihrer Arbeit berücksichtigt hat. Ich empfand Rodwins Zusammenfassungen der rechtlichen Definitionen als überaus hilfreich, um das Wesen von Interessenkonflikten besser zu verstehen und vor allem, Möglichkeiten ihrer effektiven Vermeidung und Regulierung aufzuzeigen.
Ich plädiere heute mehr als je dafür, die Regulierung von Interessenkoflikten auf eine solche traditionell-rechtliche Definition zu gründen. Der Hauptgrund ist, dass sie die Verpflichtungen/Pflichten als Schlüsselreferenz klar hervorhebt. In einem neueren Artikel drückte Professor Rodwin seine Beunruhigung darüber aus, dass die Thompson-IoM Definition Probleme schaffe, mit ihrer Beschreibung eines Interessenkonfliktes als einem Konflikt zwischen “primären“ und “sekundären“ Interessen und nicht als einem„Konflikt zwischen Verpflichtungen und Interessen“.[14] In der Tat, Verpflichtungen beziehen sich auf eine völlig andere Ebene als „sekundäre“ finanzielle Interessen.
Um Rodwin’s rechtliche Definition anwenden zu können, müssen wir die rechtlichen und grundlegenden ethischen Pflichten der Individuen präzisieren. So können wir am besten sehen, ob ein Einzelner finanzielle Verbindungen oder geteilte Loyalitäten hat, die mit diesen Pflichten in Konflikt stehen.[15]
Grundlegende ethische Prinzipien in Gesundheitsberufen sind z.B. das uralte Diktum: „Als erstes füge keinen Schaden zu“ und auch die Pflicht, verständliche Information und Rat zu geben, die voll und ganz im Interesse der Gesundheitssubjekte sind.
Mit diesem umfassenderen Konzept wäre das derzeitige Chaos in der globalen Gesundheitspolitik vielleicht vermieden worden. Zum Beispiel die Tatsache, dass es heute ein Hybrid zwischen öffentlichen und privaten Akteuren, die sogenannte Scaling-Up Nutrition (SUN) „Multi-stakeholder Plattform“ oder „Bewegung“ gibt, die ursprünglich als PP PPP – principled, peoples public-private partnership – lanciert wurde.[16] Hier werden „gemeinsame“ Belange zum Referenzpunkt von Interessenkonflikt-Regulierung erklärt und diese fragwürdige Interpretation in den Ländern verbreitet, die zu SUN-countries erklärt wurden.[17]
Es geht also um die Notwendigkeit, grundlegende rechtliche und ethische Pflichten für einzelne Angehörige der Gesundheitsberufe auszubuchstabieren? Wie würde das aussehen für Leute, die bei internationalen Organisationen arbeiten, bei der UN oder der WHO?
Ich habe bisher nur ein erstes Beispiel gegeben für weithin anerkannte und richtungsweisende ethische Prinzipien für Gesundheitspersonal. Für alle, die im internationalen Gesundheitsbereich tätig sind, gilt natürlich der Grundsatz, der als erster in der Verfassung der WHO verankert ist: es geht um den Schutz und die Förderung unser aller Menschenrecht auf Gesundheit.
Manche Personenkreise brauchen noch einen besonderen Schutz. Für diejenigen, die sich mit Kinderernährung befassen, im politischen Bereich, aber auch für KinderärztInnen und Stillberaterinnen, gelten daher zusätzliche Verpflichtungen, die sich aus den Verfassungen der UN und der WHO und aus Menschenrechtsdokumenten und relevanten Kodices und ergeben.
Die Kinderrechtskonvention der UN (CRC) fordert z.B. alle gesellschaftlichen Akteure dazu auf, immer „im besten Interesse des Kindes“ zu handeln. Sie schreibt auch „das Recht des Kindes auf einen bestmöglichen Gesundheitszustand“ fest oder die Verpflichtung „sicherzustellen, dass alle Bereiche der Gesellschaft, insbesondere Eltern […] informiert und unterstützt werden im Hinblick auf Grundwissen über Kindergesundheit und -ernährung, [und] die Vorteile des Stillens.“ (Art. 24.1 und 24.2e).[18]
Gibt es andere Gründe, eine rechtliche Definition zu bevorzugen?
Ein zweiter Grund ist, dass nicht nur Professor Rodwin, sondern inzwischen auch die Rechtsprofessorin Anne Peters empfiehlt, Loyalitäten in die Konzeptualisierung von CoI einzubeziehen. Das ist eine ihrer Schlussfolgerungen aus theoretischen Überlegungen zum Thema Interessenkonflikte in „Global Governance“.[19] Die Thompson-IoM Definition kann dazu führen, Probleme zu übersehen, die durch geteilte oder sich widersprechende Loyalitäten entstehen.
Finanzielle Interessenkonflikte sind relativ einfach zu verstehen. Sie werden durch alle möglichen Arten von Anreizen geschaffen. Die Forschung aus vielen Berufsfeldern hat gezeigt, dass finanzielle Anreize – oder Beziehungen – uns dazu verleiten können, wissentlich oder unbeabsichtigt auf eine Art zu handeln, die unsere Verpflichtungen verletzt gegenüber denjenigen, die von unserem Urteil abhängen.
Definitionen von Interessenkonflikten, die die Frage von Loyalitäten einschließen, sind etwas komplizierter. Aber sie beschreiben genauer, wie zum Beispiel Gesundheitsfachkräfte ihre Loyalität gegenüber der Person, der sie zu dienen haben, aufspalten, weil sie in potenziell sich widersprechenden Rollen stecken.
Ein viel zitiertes Beispiel ist der Rollenkonflikt von Ärzten, die als Forscher an einem neuen Arzneimittel arbeiten, während sie sich gleichzeitig um das kranke Forschungssubjekt kümmern. Auch im öffentlichen Gesundheitsbereich übernehmen Gesundheitsfachkräfte oder Behördenvertreter oft sich widersprechende Rollen. Seit das Partnerschaftsmodell sich im internationalen Gesundheitsbereich verbreitet hat, ist es nicht unüblich, dass ein und dieselbe Person Fundraising betreibt, und gleichzeitig Hauptakteur bei der Ausarbeitung von Richtlinien für die Beziehung zwischen öffentlichen Institutionen & privaten Geldgebern ist – und eventuell auch noch eingeladen wird, die politische Debatte zu beeinflussen. Ich frage mich oft, welche Folgen das bisher schon hatte.
Wenn Loyalitätsprobleme gezielter angesprochen würden, könnte man sagen, dass viele UN-Mitarbeiter in den vergangenen 20 Jahren in eine Position gespaltener Loyalität gedrängt wurden. Das Modell hinter den globalen PPPs oder „Multi-Stakeholder Partnerschaften“ (MSPs) oder „Plattformen“ fordert von ihnen, im Geist des Vertrauens“ ( „spirit of trust“ oder sogar nach einem für alle Akteure verbindlichen „principle of trust“) zu handeln und „Win-Win-Situationen“ im Interesse beider Seiten zu sichern.
Das Verständnis, dass es die Aufgabe von Amtsträgern, UN FunktionärInnen und GesundheitsexpertInnen ist, zu gewährleisten, dass der Umgang mit kommerziellen Akteuren ganz im Interesse derjenigen ist, denen gegenüber sie eine Treuepflicht haben, wurde allmählich untergraben. Auch der Grundsatz, einen gebührenden Abstand (arms-length) zwischen öffentlichen und kommerziellen Akteuren einzuhalten, ist im Zeitalter des „Verpartnerns“ verloren gegangen – ebenso die Einsicht, dass solche Interaktionen Wachsamkeit (vigilance) und nicht blindes Vertrauen erfordern.
Das hört sich alles ein bisschen kompliziert an. Gibt es eine einfachere Art, die Botschaft näher zu bringen?
Seit IBFAN mich zum ersten Mal bat, Interessenkonflikte in öffentlich-privaten „Partnerschaften“ und „Allianzen“ zu erklären, begann ich populäre Sprüche zu sammeln. Das IBFAN-Netzwerk hat Mitglieder in der ganzen Welt und so bekam ich Beispiele in vielen Sprachen, die alle zur Bebilderung der Problematik benutzt werden können: Zum Beispiel, “wes Brot ich ess, des Lied ich sing“ oder “du beißt nicht die Hand, die dich füttert“.
In Deutschland sagen wir auch: „Kleine Geschenke erhalten die Freundschaft.“ Gemeinhin ist es also bekannt, dass auch kleine Gaben ein Gefühl der Verpflichtung hinterlassen. Die Forschung hat in der Tat gezeigt, dass wir gewöhnlich das Ausmaß unterschätzen, in dem sogar geringfügige finanzielle Zuwendungen und Geschenke unsere Urteile im Interesse derjenigen verschieben, die sie machen. Firmen bauen auf das, was die Theorie des Interessenkonflikts den ‚blinden Fleck der Voreingenommenheit’ (bias blind spot) nennt.
Vor kurzem präsentierte ich auf einer IBFAN-GIFA Pressekonferenz einfache Bilder zu Interessenkonflikten in der Global Health Governance, die Interessierte im Internet ansehen können.[20] Ich ermutige die Leserinnen und Leser des Pharma-Briefes, sich auch den Beitrag von Professor David Klemperer auf dieser Pressekonferenz anzusehen. Er zitiert Schulungsmaterial von Pharmafirmen, das Pharmavertreter lehrt, bei Ärzten freundschaftliche Gefühle zu wecken, indem sie sie z.B. zum Essen einladen, dabei aber selbst nie zu vergessen, dass diese Beziehung rein kommerzieller Natur ist.
Schaut euch auch die Abbildung einer Person mit einem Spalt im Kopf an. Sie illustriert sehr gut die Tatsache, dass der Interessenkonflikt letztlich in unserem Kopf stattfindet. Das Bild macht deutlich, dass Leute von außen nicht sehen können, was in unseren Köpfen passiert, ob und in welchem Grad wir durch gewisse finanzielle Beziehungen oder gespaltene Loyalitäten beeinflusst werden.
Und das ist der Grund, warum gute Regelwerke zu Interessenkonflikten versuchen, Interessenkonflikte zu identifizieren und bestmöglich zu vermeiden, ehe Schäden verursacht werden.
Professorin Anne Peters betont, ein effektives Konzept darf die Definition nicht ausufern lassen. Ein Interessenkonflikt sei letztlich ein Konflikt im Inneren von Menschen und Institutionen, die öffentliche Interessen vertreten und nicht zwischen ihnen und kommerziellen Akteuren. Sie weist darauf hin, dass manche Theoretiker Konflikte zwischen Akteuren als widersprüchliche, konfligierende Interessen bezeichnen. Im Englischen wäre das eine Unterscheidung zwischen Conflicts of Interest (CoIs) and Conflicting Interests.
Durch diese Unterscheidung würde wieder klar, dass wir eine Debatte über zwei verschiedene Dinge führen müssten: Auf der einen Seite über die vernachlässigten Interessenkonflikte in der internationalen Arena; auf der anderen Seite darüber, dass es einen fundamentalen Unterschied gibt zwischen den Fiduciary duties – den Treuepflichen – von öffentlichen Institutionen und denen von Großunternehmen, die ihren Aktionären gegenüber die Verpflichtung haben, den Profit zu mehren. Beide Problematiken werden im neoliberalen Partnerschaftsmodell bewusst verdrängt und Kritiker mundtot gemacht.
Kannst du mir erklären, warum sich alle, die sich mit internationaler Gesundheitspolitik beschäftigen, Interessenkonflikte in den Fokus nehmen sollten?
Weil die bestmögliche Vermeidung von CoIs ein Grundpfeiler professioneller Integrität ist. Ich möchte dazu ermutigen, sich Fragen anzuschauen, die in Diskussionen um Interessenkonflikte aufkamen. Zum Beispiel hat die Forschung gezeigt, dass Ärzte, die finanzielle Zuwendungen von Pharmafirmen erhalten, dazu tendieren, zwei Dinge zu tun: Vorzugsweise das Arzneimittel der sponsernden Firma zu verschreiben, selbst wenn es nicht das Beste für eine bestimmte Krankheit ist – und zu viel zu verschreiben. Das bedeutet, sie tendieren dazu, mehr Tabletten zu verordnen als die Leute brauchen.
Ein anderes wichtiges Beispiel ist die Debatte um Muttermilchersatzprodukte. Darauf kann man sich bis heute berufen. Die WHO konnte Anfang der 1980-er Jahre noch mit Hilfe von UNICEF einen internationen Kodex für Marketingmethoden verabschieden.
Danach haben diese Firmen mit Hilfe ihrer politischen PR-Experten eine Strategie entwickelt, die andere Firmen im Gesundheitsbereich kopierten und immer weiterentwickelten, um die Verabschiedung weiterer spezifischer internationaler Verhaltenskodizes zu verhindern. Einzige Ausnahme ist die Tabak-Konvention.
Interessant ist wie in der internationalen Säuglingsnahrungsdebatte weitere relevante Resolutionen der Weltgesundheitsversammlung gezielt Interessenkonflikte ansprachen. Eine Zusammenfassung machte unlängst der UNICEF-Jurist David Clark. Diese Zusammenfassung könnte auch die Debatte von Interessenkonflikten bezüglich anderer „commerciogenic diseases“ – von kommerziellen Praktiken erzeugten Krankheiten – beleben.[21]
Wie kann man am besten über Interessenkonflikte sprechen?
Das ist nicht so einfach. Wenn ich das Thema anspreche, ist die Reaktion oft „jetzt kommt die schon wieder mit ihrer alten Leier.“
Es geht leider nicht nur um Reaktionen von PolitikerInnen, FunktionärInnen und WissenschaftlerInnen, die Gelder von der Industrie oder den großen Venture-Philanthropy Stiftungen von Gates und Ted Turner erhoffen. Viele GesundheitsaktivistInnen scheinen die Identifizierung und effektive Regulierung von Interessenkonflikten für eine Detailarbeit zu halten, die ihre schon überstrapazierten Kräfte zu sehr in Anspruch nimmt.
Ich habe auch lange gedacht, es sei zwar wichtig, immer wieder auf krasse Interessenkonflikte aufmerksam zu machen und das Bewusstsein für die allgemeine Problematik aufrechtzuerhalten, aber es sei am besten, die konkrete Ausformulierung von Regulierungs-Werken Spezialisten zu überlassen. Inzwischen musste ich miterleben, wie die WHO bei der Ausarbeitung ihres Frameworks for Interaction with non-State Actors (FENSA) und bei dem Modellvorschlag, wie mit Interessenkonflikten in nationalen Ernährungsmitteldebatten (conflict of interest tool) umgegangen werden soll, das Interessenkonflikts-Konzept bewusst uminterpretiert hat und es, trotz aller Kritik, als Konflikt zwischen Akteuren definiert.[22] Ratschläge von Aktionsgruppen und ExpertInnen, die die Probleme der WHO Konzeption kritisierten, wurden dabei bewusst ignoriert.[23],[24]
Ich habe heute dazu noch die bange Frage: Gibt es eigentlich noch genügend Spezialisten zu Interessenkonflikten, die den theoretischen und praktischen Nachholbedarf in der Behandlung der Fragen leisten können oder wollen? Wie weit beinträchtigt der Druck auf akademische Einrichtungen, Drittmittel einzuwerben, ihre Fähigkeit das neoliberale Modell und damit zusammenhängende Fragestellungen zu untersuchen und kritisieren?
Dennoch finde ich, dass es wichtig ist, nicht aufzugeben. Menschen die im Gesundheitsbereich arbeiten, ob als ÄrztInnen oder Gesundheitspersonal, ob als PolitikerInnen, Beamte oder AktivistInnen, sollten daran erinnert werden, dass die Regulierung von Interessenkonflikten einen unerlässlichen Schutz darstellt, ausschließlich im besten Interesse von denen arbeiten zu können, die ihrem Urteil vertrauen können müssen: den Patientinnen und Patienten. Letztendlich sind alle Menschen auf ein funktionierendes, bezahlbares Gesundheitssystem angewiesen.
Wir können heute nicht auf die WHO hoffen, die Probleme anzugehen?
Ja, leider. Die WHO ist, nachdem sie das Thema lange vernachlässigt hat, zur aktiven Umdefininition von Interessenkonflikten übergegangen.
Es geht nun darum, zu verhindern, dass nationale und berufständische Regulierungen zu Interessenkonflikten ebenfalls untergraben werden. Das ist besonders zu befürchten, wenn diese Akteure in Multi-Stakeholder Partnerschaften eingebunden werden.
Das muss nicht heißen, dass nun alle BürgerInnen ExpertInnen in Sachen Interessenkonflikte werden müssen. Aber es gibt ein Werkzeug aus diesem Bereich, dass jedeR von uns verwenden kann. Es heißt „reasonable person test“ – ein Test durch eine vernünftige Person, nachdem sie alle relevanten Informationen erhalten hat. Als Grundlage könnten die obengenannten OECD Leitlinien über Interessenkonflikte im Öffentlichen Dienst8 dienen, die fordern, dass öffentliche Einrichtungen gegenüber der Öffentlichkeit rechenschaftspflichtig sind (public scrutiny). Sonst gehe das Vertrauen in diese Institutionen verloren.
Dies ist eines der Schlüsselprinzipien im Umgang mit Interessenkonflikten im öffentlichem Bereich. Und da sehe ich einen Ansatzpunkt, wie jeder von uns zur Vermeidung und besseren Regulierung von Interessenkonflikten beitragen kann. Die Theorie des Interessenkonflikts besagt, dass jede Situation, die Gesundheitsfachleute oder die besorgte Öffentlichkeit als Interessenkonflikt wahrnehmen, untersucht werden muss und damit verbundene Fragen geklärt werden müssen.
Stellt z.B. eine Liste all der Situationen auf, die ihr als Interessenkonflikt wahrnehmt. Das kann helfen zu klären, welche Probleme Interessenkonflikte im rechtlichen Sinne sind, und welche andere Fragen eventuell noch angesprochen werden müssen, auch wenn sie nicht unter CoIs im engeren Sinne fallen. Dieses anerkannte OECD Prinzip könnte der derzeitigen Tendenz entgegenwirken, Menschen und Gruppen, die Interessenkonflikte oder konfligierende Interessen ansprechen, aktiv aus der Debatte auszuschließen oder ihnen die Gelder zu streichen.
Viele Angehörige der Gesundheitsberufe und öffentlich Bedienstete weisen heute darauf hin, dass es für sie schwierig ist, Interessenkonflikte bei ihrer Tätigkeit zu vermeiden, weil ihre Einrichtung in einer engen Beziehung zu kommerziellen Akteuren steht. Was könnte man dagegen machen?
Einige TheoretikerInnen behandeln dieses Problem unter der Bezeichnung „institutioneller Interessenkonflikt“,10 andere innerhalb des weiteren Rahmens „institutioneller Korruption“ (die sich von Korruption im engeren Sinne unterscheidet).[25],[26]
Es ist unfair und unrealistisch, zu erwarten, dass Individuen Interessenkonflikte komplett vermeiden können, so lange nichts gegen die Ursachen unternommen wird, die dafür sorgen, dass solche Konflikte exponenziell zunehmen. Es ist höchste Zeit, an die Politik heranzutreten, um zu einer wirksamen Regelung von Interessenkonflikten im Bereich der internationalen Gesundheitspolitik zu kommen..
Die wichtigste Ursache für die Zunahme von Interessenkonflikten in der internationalen Gesundheitsarena ist die Hegemonie des Public-Private-Partnerschafts-Modells. Es ist ein Teil der neoliberalen Umstrukturierung unserer Gedankenwelt und Institutionen, die seit Ende der 1980er Jahre weltweit stattgefunden hat. Eine breitere öffentliche Debatte über Interessenkonflikte kann weitere Probleme in diesem Zusammenhang ansprechen. Sie könnte Forderungen nach lange vernachlässigten Instrumenten wie „cooling off periods“ für öffentlich Bedienstete, bevor sie in den Privatsektor wechseln und umgekehrt, dem Schutz von Whistle-blowers und dem Zugang zu wichtigen Informationen im Sinne des amerikanischen Freedom of Information Acts neuen Aufwind verleihen.
Eine solche Debatte würde auch die Absurdität und Gefahr der Idee enthüllen, Großfirmen zu „Richtern in eigener Sache“ zu machen, das heißt, sie z.B. als Partner in die Regulierung ihrer eigenen problematischen Geschäfts- und politischen Praktiken einzubinden – eine Praxis, die sicher entscheidend dazu beigetragen hat, eine wirksame internationale Regulierung der Vermarktung dickmachender Produkte seit 2004 zu verschleppen.
Das Interview führten Claudia Jenkes und Jörg Schaaber.
Artikel aus dem Pharma-Brief 9/2019, S.2
Bild © Lida Lhotski
[2] Richter J (1999) Sponsorship as corporate engineering of consent strategy. In: The ties that bind? Weighing the risks and benefits of pharmaceutical industry sponsorship. Amsterdam: Health Action International (HAI Europe)
[3] Richter J (2001) Holding corporations accountable: Corporate conduct, international codes, and citizen action. London and New York: Zed Books. Für Briefing paper, basierend auf Buch www.thecornerhouse.org.uk/sites/thecornerhouse.org.uk/files/26codes.pdf
[4] Ollila E (2003). Global-health related public-private partnerships and the United Nations, Globalism and Social Policy Programme (GASPP), University of Sheffield., 8 pp. http://praha.vupsv.cz/fulltext/ul_303_2.pdf
[5] Richter J (2004) Public-private partnerships and international health policy making: How can public interests be safeguarded? Helsinki: Ministry for Foreign Affairs of Finland, Development Policy Information Unit www.webcitation.org/query.php?=http://global.finland.fi/julkai/pdf/public_private2004.pdf&refdoi=10.1186/1744-8603-1-6 Kurzfassung Public-private partnerships and Health for All How can WHO safeguard public interests? siehe www.aaci-india.org/Resources/Public-Private-Partnerships-and-Health-for-All.pdf
[6] WHO/UNICEF (2003) Global strategy for infant and young child feeding. Geneva, World Health Organization www.who.int/maternal_child_adolescent/documents/9241562218/en/ , publications
[7] Richter J (2005) Conflicts of interest and policy implementation: reflections from the fields of health and infant feeding. Geneva, IBFAN-GIFA, 2nd ed. www.aaci-india.org/Resources/Conflicts-of-Interest-and-Policy-Implementation-judith-ritcher.pdf
[8] OECD (2003) OECD Guidelines for Managing Conflict of Interest in the Public Service in OECD (2005) Managing Conflict of Interest in the Public Sector. Paris: OECD, p 95-110 www.oecd.org/gov/ethics/managing-conflict-of-interest-in-the-public-sector-9789264018242-en.htm
[9] Thompson, DF (1993) Understanding Financial Conflicts of Interest. N Engl J Med 329, p 573 http://med.stanford.edu/content/dam/sm/bioethics/resources-secure/Thompson2006NEJM.pdf
[10] Lo B and Field MJ (editors) (2009) Institute of Medicine (US) Committee on Conflict of Interest in Medical Research, Education and Practice. Conflict of interest in medical research, education and practice. Washington DC, National Academics Press. www.ncbi.nlm.nih.gov/books/N
[11] Lieb et al. (2018) Interessenkonflikte, Korruption und Compliance im Gesundheitswesen. Berlin: Medizinisch Wissenschaftliche Verlagsgesellschaft.
[12] Rodwin MA (1993) Medicines, Money & Morals: Physicians‘ Conflicts of Interest. Oxford: Oxford University Press
[13] Rodwin MA (2017) Attempts to redefine conflicts of interest. Legal Studies Research Paper Series. Research Paper, Suffolk University Law School. 7 December https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3084307
[14] Rodwin MA (2017) Attempts to redefine conflicts of interest. Legal Studies Research Paper Series. Research Paper, Suffolk University Law School. 7 December https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3084307
[15] Beispiel einer Checkliste für Interessenkonflikte: Rodwin M (2019) Conflict of Interest in the Pharmaceutical Sector: A Guide to Public Management. Legal Studies Research Paper Series(Research Paper 19-3) https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3317603
[16] Lhotska L et al. (2012) Conflicts of Interest and Human Rights-Based Policy Making: the Case of Mater-nal, Infant, and Young Children’s Health and Nutrition, Right to Food and Nutrition Watch, Issue 7, p 31-36 www.righttofoodandnutrition.org/files/R_t_F_a_N_Watch_2012_eng.pdf Zusammenfassung unter www.fian.org/fileadmin/media/publications_2015/FIAN_Right_to_Food_Journal_Vol_7_No._1.pdf p 8
[17] Richter J (2015) Conflicts of interest and global health and nutrition governance - The illusion of robust principles. BMJ, RR, 12 February. www.bmj.com/content/349/bmj.g5457/rr
[18] UNICEF (1989/90) United Nations Convention for the Rights of the Child (CRC) www.unicef.org/child-rights-convention/convention-text
[19] Peters A (2012) Conflict of interest as a cross-cutting problem of governance & Conclusion. In: Peters & Handschin. Conflicts of interest in global, public and corporate governance. Cambridge & New York: Cambridge University Press: S. 3-38 & 357-421
[20] IBFAN-GIFA (2018) Health governance in the public interest? WHO redefines conflicts of interest and risks undermining public health mandates. Press Conference, Geneva, Geneva Press Club/Club Suisse de la Presse. www.gifa.org/press-conference-health-governance-in-the-public-interest-who-redefines-conflicts-of-interest-and-risks-undermining-public-health-mandates (hier finden sich auch ausführlichere Powerpoint-Präsentationen)
[21] Clark D (2017) Avoiding Conflict of Interest in the in the field of Infant and Young Child Feeding: better late than never. World Nutrition; 8
[22] Richter J (2015) Time to debate WHO’s understanding of conflicts of interest. BMJ RR (22 October) www.bmj.com/content/348/bmj.g3351/rr
[23] Lhotska L and Gupta A (2016) Whose health? The crucial negotiations for the World Health Organization’s future. APPS Policy Forum. www.policyforum.net/whose-health
[24] Richter J (2017) Comments on Draft Approach for the prevention and management of conflicts of interest in the policy development and implementation of nutrition programmes at country level. www.who.int/nutrition/consultation-doi/judith_richter.pdf
[25] JLME Special Issue (2013) Institutional Corruption and the Pharmaceutical Industry. Journal of Law Medicine and Ethics (JLME) https://papers.ssrn.com/sol3/papers.cfm?abstract_id=2298140
[26] Marks J (2019) The Perils of Partnership: Industry Influence, Institutional Integrity, and Public Health USA.Oxford: Oxford University Press
Weiteres relevantes Material
IBFAN-GIFA www.gifa.org/international/conflits-dinterets
Publikationen von Judith Richter: www.ibme.uzh.ch/en/Biomedical-Ethics/Team/Affiliates/judithrichter.html
Interessengruppen auf den Leim gegangen?
Projekt des Gesundheitsministeriums trifft auf breite Kritik
Am 19. Februar wurde der Global Health Hub Germany offiziell der Öffentlichkeit präsentiert. Teile der Zivilgesellschaft blieben der Veranstaltung, die viele Fragen offen ließ, fern. Zentrale Aspekte sind nach wie vor unklar, etliche KritikerInnen sehen weiterhin keinen Bedarf für dieses neue Gremium.
Mit einer gut dreistündigen Veranstaltung eröffnete das Bundesministerium für Gesundheit (BMG) am 19. Februar in Berlin den so genannten Global Health Hub Germany (GHHG). Das vom Ministerium geförderte neue Gremium soll nach eigenem Bekunden Akteure aus Politik, Zivilgesellschaft, Wissenschaft und Wirtschaft zusammenbringen, um die nachhaltigen Entwicklungsziele der Vereinten Nationen im Bereich Gesundheit und Partnerschaften voranzubringen.
Schon im Vorfeld hatten allerdings etliche Organisationen umfangreiche Kritik an der Plattform und ihrer Entstehung geäußert (wir berichteten [1],[2]). Sie bemängelten, dass das Projekt von der Wirtschaft lanciert und in einem wenig transparenten Prozess gegen Vorbehalte anderer Ministerien sowie von Teilen der Zivilgesellschaft durchgedrückt worden sei.
Bleibende Baustellen
Einige Organisationen, darunter auch die BUKO Pharma-Kampagne, blieben den Feierlichkeiten aus Protest fern. Gemeinsam mit der Deutschen Plattform globale Gesundheit veröffentlichten wir eine Pressemitteilung , die den inhärenten Konflikt des Gremiums zwischen Gemeinwohlinteressen und Gewinninteressen im Gesundheitsbereich hervorhob: „Wenn ein Global Health Hub etwas erreichen soll, darf er nicht den Interessen der Wirtschaft Vorrang einräumen.“ [3]
Angesichts der Kontroversen um den Hub ist es befremdlich, dass in dem Event kein Platz für kritische Stimmen aus dem Plenum vorgesehen war. Als Agendapunkt fand sich zwar eine „Live-Umfrage mit Teilnehmern“ (sic!) – die dafür veranschlagte Zeit betrug jedoch ganze fünf Minuten und TeilnehmerInnen konnten lediglich digital auf wenige ausgewählte Fragen antworten. Wirklich negative Optionen waren nicht vorgesehen.
Der Zivilgesellschaft wurde unter dem Punkt „Stimmen der Akteursgruppen“ immerhin ein kurzes Statement eingeräumt. Das vorgegebene Zeitfenster: zwei Minuten. Bemängelt wurde in dem Beitrag von Gisela Schneider (Difäm), es bleibe nach wie vor unklar, was das genaue Ziel des GHHG sein solle. Zudem seien zentrale Fragen, etwa zur Leitung und Lenkung, Mitgliederauswahl oder Themen-Selektion weiterhin ungeklärt. Entsprechende Infos bleibt auch der sechsseitige Flyer auf der neuen Website des GHHG schuldig.[4]
Heike Baehrens (MdB) zeigte sich auf dem Podium irritiert, dass weder das Bundesministerium für wirtschaftliche Zusammenarbeit und Entwicklung (BMZ) noch das Bundeministerium für Bildung und Forschung (BMBF) prominent vertreten waren. Überraschend kam dies allerdings nicht, hatte das BMG doch bei seinem Vorstoß in der Vergangenheit beide Ressorts zunächst mehr oder minder übergangen. Sowohl der Vernetzungsgedanke, den der Hub für sich selbst postuliert, aber auch eine nachhaltige, kohärente Politik wurden hier lediglich parodiert.
Vorgeben statt anhören
Bemerkenswert auch die Themensetzung. Bundesgesundheitsminister Jens Spahn präsentierte beim Launch des GHHG vier Arbeitsfelder, um die sich der Hub zunächst kümmern wird: Antibiotika-Resistenzen, Digitalisierung, Krebsbekämpfung und Tropenkrankheiten.[5] Diese Auswahl erfolgte wohlgemerkt nicht über ein Votum der Mitglieder des Hubs, die es zum Zeitpunkt der Gründung ja noch gar nicht geben konnte.
Die Vorgehensweise ist symptomatisch für die paradoxe Vorgehensweise des Hubs. Anstatt in einem ersten Schritt Meinungen zu den Prioritäten anzuhören, sind die Eckpfeiler von einer Gruppe undurchsichtiger Zusammensetzung schon vorab eingeschlagen.
Ein zweites Beispiel zeigt ebenfalls den frappierenden Gegensatz zwischen formuliertem Anspruch und Realität: Selbst die Key-Note mit dem plakativen Titel „Warum braucht es einen Global Health Hub Germany?“ konnte letztlich keine befriedigende Antwort auf eben jene Frage geben. „Wir müssen untypische Partnerschaften fördern, damit die Global Health Familie größer wird“, so die Vortragende Frau Ilona Kickbusch (Global Health Centre).[6] Unspezifischer und inhaltsleerer geht es kaum. Durch die gewählte Terminologie „Familie“ werden real existierende Interessengegensätze zugekleistert und unkritisch Public Private Partnerships propagiert.
Im Vergleich zu bereits bestehenden Strukturen ist der GHHG in seiner jetzigen Form kein innovativer Ansatz. Die NGOs sind gut vernetzt und sprechen auch mit Politik, Wissenschaft, Gewerkschaften und Unternehmen. Letztlich liegt in der hoch problematischen Genese der Hase im Pfeffer. Es sei daran erinnert, dass der GHHG konzipiert wurde, ohne dass zuvor alle eingeplanten Akteursgruppen überhaupt im Rahmen einer Bedarfsanalyse zur Notwendigkeit Gehör fanden. Die Zivilgesellschaft erfuhr erst spät und auf Umwegen von dem Projekt. Wirtschaft und philanthropische Stiftungen hingegen hatten den Prozess von Beginn an geprägt. Bedarf wurde also offenbar vor allem von diesen Gruppierungen angemeldet.
Was vom Abend übrig blieb
Die während des Launch verkündeten ersten Themenschwerpunkte der Plattform (s.o.) verweisen dann auch entsprechend deutlich auf diese eingebaute Schräglage. So bietet beispielsweise gerade der Bereich Digitalisierung für Akteure der Wirtschaft ein äußerst lukratives Betätigungsfeld.
Die von Bundesminister Spahn auf der Veranstaltung verkündete finanzielle Unterstützung eines WHO-Programms zu vernachlässigten Tropenkrankheiten fand ihren Widerhall in der direkten Ankündigung der Gates-Stiftung, ebenfalls Förderung in Millionen-Höhe beizusteuern. Von glücklichen Zufällen mag man hier nicht ausgehen.
Es steht zu befürchten, dass der Hub als Vehikel von Eigeninteressen der Beteiligten auch andere Prozesse zu globaler Gesundheit beeinflussen könnte, etwa die neue globale Gesundheitsstrategie der Bundesregierung. Dies gilt umso mehr, sollte sich der Hub im Laufe der Zeit tatsächlich zum Hauptanlaufpunkt für Themen globaler Gesundheit entwickeln und damit eine wichtige Gatekeeper-Funktion übernehmen. Akteure der Zivilgesellschaft sollten sich also ernsthaft überlegen, ob ihre Beteiligung am GHHG am Ende nicht vor allem als Feigenblatt dient.
Eine ehrliche und konstruktive Debatte, gerade zu einem so elementaren Thema wie Global Health, muss Kontroversen aushalten können, braucht sie letztlich sogar zwingend. Die neue Plattform des BMG scheint gerade das nicht zu bieten. Sie ist eher darauf ausgerichtet, den kritischen Diskurs einzuhegen und einer bereits vorab festgelegten Agenda zu folgen, statt einen transparenten und fairen Austausch mit Mehrwert zu stimulieren. Inwieweit dies von politischer Seite so intendiert war oder man hier den Interessen anderer auf den Leim gegangen ist, stellt eine der vielen offenen Fragen dar. (MK)
Artikel aus dem Pharma-Brief 1/2019, S. 1
[1] Pharma-Brief (2018) Bundesregierung hört zu. Nr. 7, S. 1
[2] Pharma-Brief (2018) Abgekartetes Spiel. Nr. 8-9, S. 1
[3] Deutsche Plattform für Globale Gesundheit (2019) Falsche Weichenstellung. Pressemitteilung 19. Feb. www.plattformglobalegesundheit.de/falsche-weichenstellung/ [Zugriff 19.02.2019]
[4] Global Health Hub Germany (2019) Menschen & Ideen zusammenbringen. Perspektiven erweitern. Themen setzen. www.globalhealthhub.de/#2 [Zugriff 20.02.2019]
[5] Bundesministerium für Gesundheit (2019) Bundesgesundheitsminister Jens Spahn: „Starkes deutsches Netzwerk für globale Gesundheit“. Pressemitteilung 19.02.2019. https://www.bundesgesundheitsministerium.de/presse/pressemitteilungen/2019/1-quartal/global-health-hub-germany.html [Zugriff 20.02.2019]
[6] Deutsches Ärzteblatt (2019) Bundesgesundheitsministerium fördert Netzwerk für Globale Gesundheit. www.aerzteblatt.de/nachrichten/101233 [Zugriff 21.02.2019]
Global gesunde Geschäfte
German Health Alliance der Wirtschaft
Die deutsche Wirtschaft will die globale Gesundheit stärken, so die Eigenwerbung der neu gegründeten „German Health Alliance“.[1] NGOs und staatliche Träger der Entwicklungshilfe sind mit im Boot. Wem nützt dieses „Public Private Partnership“ (PPP)?
Eigentlich ist die vom Bundesverband der deutschen Industrie (BDI) gegründete „German Health Alliance“ (GHA) nicht ganz frisch. Sie bündelt die Aktivitäten des BDI aus drei schon länger bestehenden Exportförder-Partnerschaften, darunter dem German Healthcare Partnership (GHP).[2] Letzteres war wiederum Ideengeber für den „Global Health Hub Germany“,
der vergangenes Jahr formal vom Bundesgesundheitsministerium ins Leben gerufen wurde (wir berichteten [3],[4]).
Etwas verwirrt über die vielen „Partnerschaften“? Dahinter könnte Methode stecken: Es kann gar nicht genug Foren geben, in denen man seine Meinung zu Gehör bringt. Um dem Ganzen mehr Legitimität zu verleihen, holt man sich einige NGOs an Bord und verstrickt Bundestagsabgeordnete – ein paar WissenschaftlerInnen als Garnitur machen sich auch nicht schlecht.
Die Industrie versucht mit aller Macht, den Diskurs über globale Gesundheit in ihr genehme Bahnen zu lenken. Die „German Health Alliance“ wird am 26.11.2019 aus der Taufe gehoben. Thema der Veranstaltung: „Deutschlands Rolle und Verantwortung in Globaler Gesundheit“. Als Ziel wird ausdrücklich genannt, „die Zusammenarbeit zu stärken und gemeinsam neue Wege zu beschreiten.“[5]
Es geht nicht nur um Exportförderung der deutschen Pharma- und Medizintechnikindustrie, sondern auch um möglichst günstige Rahmenbedingungen für die deutsche Wirtschaft. Durch den UN-Beschluss, Gesundheitsversorgung für alle (UHC[6]) hoch auf die politische Agenda zu setzen, locken größere Märkte.
Angesichts der massiven Kritik an hohen Preisen für Medikamente weltweit gilt es, eine harte Regulierung der Geschäfte zu verhindern. Und was hilft da besser, als möglichst viele AkteurInnen an den Tisch zu holen, um sie für die eigenen Zwecke einzuspannen. Und selbst wenn das nicht gelingt, hält man die Leute so beschäftigt.
Die GHA rühmt sich auch der engen Kooperation mit dem World Health Summit.[7] Dieses industrielastige jährliche Treffen zu globaler Gesundheit wird von Prof. Detlev Ganten, Vorsitzender des Stiftungsrats der Charité, geleitet. Was liegt da näher, als dass die GHA ihm am 26.11. auch den „German Global Health Award“ verleiht? Überreicht wird der Preis von Gesundheitsminister Jens Spahn. Die Laudatio hält Prof. Karl Max Einhäupl, ehemaliger Vorstandsvorsitzender, Charité. Ganten ist übrigens auch im Advisory Board der GHA – so schließen sich die Kreise. (JS)
Artikel aus dem Pharma-Brief 9/2019, S.1
[1] https://gha.health/ [Zugriff 19.11.2019]
[2] Die anderen sind German Healthcare Export Group (GHE) und die German-Sino Healthcare Group (GSHCG)
[3] Pharma-Brief (2018) Abgekartetes Spiel. Nr. 8/9, S. 1
[4] Pharma-Brief (2019) Gesundheitshub krankt. Nr. 4/5, S. 4
[5] GHA (2019) https://gha.health/wp-content/uploads/Programm_26112019_deutsch.pdf [Zugriff 19.11.2019]
[6] Universal Health Coverage
[7] https://ghp-initiative.de/about-us/ [Zugriff 20.11.2019]
Gesundheitshub krankt
Uneinigkeit bei Struktur und Mitbestimmung
Der Global Health Hub Germany (GHHG) bleibt ein Problemprojekt des Bundeministeriums für Gesundheit (BMG). Denn das Hauruck-Verfahren bei seiner Gründung rächt sich nun bei der Diskussion um die interne Entscheidungskultur. Obwohl der Hub schon Aktivitäten entfaltet, gibt es keine Satzung, die die Rechte von Mitgliedern und Lenkungskreis regelt.
Nach anhaltender externer Kritik, zuletzt etwa in einem Beitrag in welt-sichten,[1] rumort es mittlerweile vernehmlich in den „Eingeweiden“ des Hubs. Ursächlich dafür sind vor allem auch die Versäumnisse beim Aufbau des Prestige-Objekts (wir berichteten[2]).
Entzündet haben sich die Diskussionen innerhalb des GHHG vor allem an der Frage, wie die noch ausstehende Satzung aussehen soll und dabei besonders, in welchem Verfahren sie bearbeitet und verabschiedet werden wird. In einem Brief an das BMG und die Gesellschaft für Internationale Zusammenarbeit (GIZ) benennt Eva-Maria Schreiber, Bundestagsabgeordnete der Linken und Mitglied im Hub, das Problem:[3] Demnach sei ursprünglich zugesagt worden, die Satzung in einem transparenten Konsultationsprozess gemeinsam mit allen Mitgliedern zu erarbeiten – nun solle jedoch wohl zeitnah der Lenkungskreis allein eine finale Version verabschieden. Die GIZ bestreitet das. Mitglieder könnten bald im Intranet des Hubs Rückmeldungen zu der von GIZ und Lenkungskreis entworfene Satzung geben.
Dennoch stellen sich (mindestens) zwei kritische Frage. Zum Einen, auf welcher Grundlage der Interims-Lenkungskreis bei diesem Prozess eigentlich handeln kann. Zum Anderen, welche Rolle denn die Mitglieder des Hubs letztlich spielen, sofern basisdemokratisches Handeln wirklich angestrebt wird.
Henne, Hub und Ei
 Durch das Drängen des BMG, den GHHG möglichst schnell öffentlichkeitswirksam auftreten zu lassen, mangelt es ihm intern grundlegend an Verfahrensregeln. Dem wohlgemerkt extern bestimmten Interims-Lenkungskreis wurde nun unter Einbezug der nicht im Hub vertretenen GIZ die Rolle zugeschoben, diesen Schwachpunkt zu beheben. Der erster Entwurf für eine Satzung kam offenbar von der GIZ, die als Auftragnehmer des BMGs die Hub-Geschäftsstelle führt, dieser wurde dann vom Interims-Lenkungskreis diskutiert. Laut früherer Aussage des GHHG wird der Interims-Lenkungskreis auch schlussendlich die Kommentierung und Verabschiedung des Satzungsentwurfs vornehmen.[4]
Durch das Drängen des BMG, den GHHG möglichst schnell öffentlichkeitswirksam auftreten zu lassen, mangelt es ihm intern grundlegend an Verfahrensregeln. Dem wohlgemerkt extern bestimmten Interims-Lenkungskreis wurde nun unter Einbezug der nicht im Hub vertretenen GIZ die Rolle zugeschoben, diesen Schwachpunkt zu beheben. Der erster Entwurf für eine Satzung kam offenbar von der GIZ, die als Auftragnehmer des BMGs die Hub-Geschäftsstelle führt, dieser wurde dann vom Interims-Lenkungskreis diskutiert. Laut früherer Aussage des GHHG wird der Interims-Lenkungskreis auch schlussendlich die Kommentierung und Verabschiedung des Satzungsentwurfs vornehmen.[4]
Dabei wird er sich allerdings nicht auf transparente und faire Verfahrensweisen stützen können – diese werden ja erst in einer Satzung festgeschrieben. Die Auflösung der existenziellen Frage, ob erst die Henne oder das Ei kam, soll offenbar wie folgt „gelöst“ werden: Die Henne legt einfach das Ei, aus dem sie dann schlüpft. Leidtragende ist hier die breite Mitgliederschaft, der eine umfangreiche Mitbestimmung in Aussicht gestellt worden war.
Kommt alles Gute von oben?
Im April kommunizierte das BMG, der Hub habe bereits fast 170 Mitglieder. Die blieben bisher allerdings weitgehend ohne direkten Einfluss, konnten lediglich beim so genannten Global Health Talk Mitte Juni erste Arbeitsgruppen gründen.[5]
Die übergeordneten Schwerpunktthemen hatte das BMG vorsorglich schon beim Start des Hub selbst festgelegt.[6] Auch der erste Lenkungskreis war vorab bestimmt worden. Es hat also schon eine gewisse Tradition, dass die Mitglieder bei den fundamentalen Entscheidungen nur begrenzt mitreden können. Momentan deutet wenig darauf hin, dass sich dies bei Ausarbeitung und Annahme der Satzung ändern könnte. Denn selbst wenn irgendwann noch digitale Rückmeldungen aufgenommen werden, ist momentan völlig unklar, ob und wie genau diese berücksichtigt werden.
Die Möglichkeit einer zeitnahen, ersten Mitgliederversammlung mit Debatte und Abstimmung über einen Satzungsentwurf, der schließlich elementar für die künftige Ausrichtung und Arbeit des Hub ist, wurde offenbar gar nicht ernsthaft erwogen. Für ein Projekt, bei dem Partizipation angeblich groß geschrieben werden sollte, ist dieses Zwischenzeugnis verheerend und wachsender Unmut verständlich. Es steht zu erwarten, dass es für den GHHG noch ein heißer Sommer wird. (MK)
Artikel aus dem Pharma-Brief 4-5/2019, S.4
Bild Henne bunt © luna4 /iStock
[1] welt-sichten (2019) Hype um den Hub. www.welt-sichten.org/artikel/36147/hype-um-den-hub [Zugriff 23.07.2019]
[2] Pharma-Brief (2019) Neues aus dem Nebel. Nr. 2, S. 4-5.
[3] Der Brief ist einsehbar unter https://www.eva-maria-schreiber.de/de/article/217.brief-an-giz-konsultationsprozess-zur-satzung-des-global-health-hubs.html [Zugriff 24.07.2019]
[4] Global Health Hub Germany (2019) Auftaktsitzung Interims-Lenkungskreis Global Health Hub Germany. www.globalhealthhub.de/de/news/auftaktsitzung-interims-lenkungskreis-global-health-hub-germany [Zugriff 23.07.2019]
[5] Global Health Hub Germany (2019) #GlobalHealthTalk2019 – Auftakt für die akteursübergreifende Zusammenarbeit des Global Health Hub Germany. www.globalhealthhub.de/de/news/globalhealthtalk2019-auftakt-fuer-die-akteursuebergreifende-zusammenarbeit-des-global-health [Zugriff 23.07.2019]
[6] Pharma-Brief (2019) Interessengruppen auf den Leim gegangen? Nr. 1, S. 1
Europäische Nutzenbewertung
Wie geht es weiter?
Über den Gesetzentwurf der EU-Kommission für eine einheitliche Nutzenbewertung von Arzneimitteln haben wir wiederholt berichtet.[1],[2],[3] Parlament und Ministerrat sind sich nicht einig. Wie geht es nach der Europawahl weiter?
Auch wenn dem wirtschaftsfreundlichen Entwurf der Kommission durch intensive Öffentlichkeitsarbeit von kritischen NGOs und Proteste der Fachwelt die meisten Giftzähne gezogen wurden, bleiben nach dem im Oktober 2018 vom EU-Parlament beschlossenen Entwurf Fragezeichen.
Abgeordnete wollen gehört werden
Das Parlament hat im Februar 2019 nochmals seinen Standpunkt bekräftigt.[4] Es möchte, dass auch nach der Europawahl auf Basis seines Beschlusses weiterverhandelt wird. Das ist im Gegensatz zum Bundestag, wo vor einer Wahl nicht abgeschlossene Gesetzgebungsverfahren komplett neu gestartet werden müssen, in der EU möglich. Die EU-Abgeordneten warnten mit ihrem Beschluss die Kommission davor, das Gesetz ohne vorherige Konsultation des Parlaments umzuschreiben.
Der Ministerrat hatte im November 2018 noch einmal strittige Punkte festgehalten.[5] Im Kern geht es um die Möglichkeit, nach wie vor nationale Entscheidungen über die Erstattungsfähigkeit von Arzneimitteln treffen zu können. „Eine große Mehrheit der Delegationen vertritt die Auffassung, dass – falls erforderlich – auch nationale klinische Bewertungen möglich sein müssen.“ Damit befinden sich die Mitgliedsstaaten im Widerspruch zu der Auffassung des Parlaments. Außerdem mahnten die Staaten an, dass eine gemeinsame europäische Nutzenbewertung „mindestens so gut“ wie nationale Bewertungen sein müssen. Das Verfahren müsste maximal transparent gestaltet werden und „strikte Regeln für Interessenkonflikte“ eingefügt werden, „um einen unabhängigen Bewertungsprozess zu garantieren.“
Da sich Parlament, Kommission und Ministerrat über den Entwurf einigen müssen, bleibt es auch nach der Europawahl spannend. (JS)
Artikel aus dem Pharma-Brief 2/2019, S. 2
[1] Pharma-Brief (2018) Wunschkonzert für Hersteller. Nr. 3, S. 1
[2] Pharma-Brief (2018) Zwischen Kommerz und Transparenz. Nr. 6, S. 5
[3] Pharma-Brief (2018) EU-HTA Update 2. Nr. 7, S. 6
[4] Europäisches Parlament (2019) Bewertung von Gesundheitstechnologien. P8_TA-PROV (2019)0120. Legislative Entschließung 14. Feb. 2019
[5] Council of the European Union (2018) Interinstitutional File 2018/0018 (COD) 14694/18, 30 Nov 2018
Essenzielle Probleme
Listen unentbehrlicher Arzneimittel sind oft nicht kohärent
Seit über 40 Jahren gibt es die Modelliste für unentbehrliche Arzneimittel der Weltgesundheitsorganisation (WHO). Die meisten Länder der Welt nutzen angepasste nationale Listen für ihre Gesundheitsversorgung. Doch die Auswahl der Wirkstoffe scheint nicht immer optimal. Und die hohen Preise für viele neue Präparate stellen ein ernstes Zugangshindernis dar.
Eine sinnvolle Auswahl an Medikamenten zu treffen, die in einem Gesundheitssystem verfügbar sein sollen, gilt international als Eckpfeiler für eine gute Versorgung. Der Zugang zu unentbehrlichen Arzneimitteln ist als Ziel in den Nachhaltigen Entwicklungszielen (SDGs) der Vereinten Nationen verankert. Nur wenige Länder – darunter auch Deutschland – leisten sich den Luxus, ohne Rücksicht auf den Nutzen (fast) alle Medikamente zu erstatten.
Die alle zwei Jahre aktualisierte Liste unentbehrlicher Arzneimittel der WHO ist ausdrücklich als Modellliste konzipiert. Sie bietet einen Orientierungsrahmen für nationale Listen. Länder sollen sie nach ihren lokalen Bedürfnissen (und finanziellen Möglichkeiten) ergänzen und nicht benötigte Mittel streichen. Auffällig sind aber die extremen Unterschiede bei den Listen, die sich nicht allein durch unterschiedliche Krankheitslast und Budgetbeschränkungen erklären lassen.
ForscherInnen von zwei öffentlichen Einrichtungen in Kanada und Großbritannien[1] haben gemeinsam mit MitarbeiterInnen der WHO die 137 vorhandenen nationalen Listen unter die Lupe genommen.[2] Auf den Listen fanden sich zusammengenommen 2.068 verschiedene Wirkstoffe. Davon tauchten allerdings 1.248 nur in zehn oder weniger nationalen Listen auf.
Die Spannbreite der Wirkstoffe ist groß, von 44 bis fast 1.000 Medikamente umfassen die Listen je nach Land. Dabei sind sie in ärmeren Ländern oft kürzer, es lässt sich aber kein eindeutiger Zusammenhang mit dem Bruttosozialprodukt erkennen. Spitzenreiter ist die Slowakei mit 983 Medikamenten auf ihrer Liste, dicht gefolgt von Syrien mit 964. Das reiche Schweden kommt hingegen mit 289 Wirkstoffen aus.
Auffälliger noch sind die Abweichungen von den 414 Wirkstoffen auf der WHO-Modellliste von 2017.[3],[4] 73 Wirkstoffe fanden sich nur auf 20% aller nationalen Listen wieder. Dagegen fanden sich rund 100 Medikamente auf fast allen Listen (>80%) wieder. Einige Listen weisen eine hohe Ähnlichkeit mit der WHO-Liste auf, d.h. die meisten der für die nationale Liste gewählten Wirkstoffe stehen auch auf der WHO-Liste, die Zahl der zusätzlichen aufgeführten Arzneimittel ist gering. Die höchste Übereinstimmung weist Pakistan mit 93% auf: Von den 373 Medikamenten auf der nationalen Liste finden sich 347 auch in der WHO-Modellliste.
Andererseits gibt es auch eine Vielzahl von Listen mit einer nur geringen Überschneidung mit dem WHO-Modell. Das liegt oft auch an der großen Zahl zusätzlicher Medikamente, die in die nationale Liste aufgenommen wurden. So finden sich von den 707 Medikamenten auf der äthiopischen Liste nur 319 (45%) auf der WHO-Liste wieder. Stärker ist die Abweichung in der oben schon erwähnten langen syrischen Liste, sie stimmt nur zu 32% mit der Modellliste überein.
Ein weiterer Schwachpunkt sind fehlende Aktualisierungen vieler nationaler Listen: Nicht wenige sind bis zu zehn Jahre alt.
Überprüfung notwendig
Die AutorInnen regen eine kritische Überprüfung der Listen auf nationaler Ebene an. Sämtliche Ergebnisse ihrer Untersuchung haben sie in einer leicht durchsuchbaren Datenbank hinterlegt.[5] Als Warnsignal sehen sie Wirkstoffe, die sich weltweit nur auf wenigen Listen finden. Ein Beispiel dafür ist Acarbose, ein sehr wahrscheinlich unwirksames Mittel gegen Diabetes,[6] das in 21 Ländern gelistet ist.[4]
Eine Schwierigkeit scheint die unzureichende Evidenzbasis für etliche nationale Entscheidungen zu sein. Allerdings ist hier die WHO-Modellliste auch kein gutes Vorbild. Die Auswahl basiert weitgehend auf einem ExpertInnenkonsens. Das Selektionsverfahren fällt weit hinter die heute üblichen Standards der Nutzenbewertung von Arzneimitteln zurück, die in vielen Industrieländern angewandt werden.
Preise als Problem
In die neueste WHO-Liste von 2019 wurden zehn neue Krebsmedikamente aufgenommen.[7] Bereits Industrieländer haben mit den hohen Kosten für diese Medikamente zu kämpfen, die Jahrestherapiekosten liegen oft über 50.000 €. Umso wichtiger ist es, dass diese Medikamente in ärmeren Ländern preisgünstig zur Verfügung stehen. Das ist aber oft nicht der Fall und häufig auch ein Grund, warum Medikamente nicht auf die nationalen Listen unentbehrlicher Arzneimittel kommen. Als Beispiel nennen VertreterInnen von NGOs und WissenschaftlerInnen den Wirkstoff Lenalidomid, der gegen das multiple Myelom eingesetzt wird und dieses Jahr auf die WHO-Liste kam.[8] Bis 2016 war Lenalidomid in Südafrika als Generikum verfügbar, die Jahrestherapiekosten betrugen pro PatientIn 2.289 US$. Dann registrierte Celgene sein Markenpräparat in Südafrika, seitdem kostet es 51.000 US$ pro PatientIn und ist deshalb im öffentlichen Gesundheitssektor, der 84% der Bevölkerung versorgt, nicht mehr verfügbar. Die Herstellungskosten für den Wirkstoff werden auf 2,55 US$ für eine Monatsbehandlung geschätzt. In Indien wurde der Patentantrag für Lenalidomid abgelehnt, dort ist das Präparat für rund 2.000 US$ verfügbar – ein Preis, der für viele InderInnen außerhalb ihrer Möglichkeiten liegt.
Das von der Industrie oft vorgebrachte Argument, dass die Forschungskosten die hohen Preise rechtfertigen, ist aus zwei Gründen nicht stichhaltig: Erstens sind PatientInnen, die sich die Medikamente aus Preisgründen nicht leisten können, sowieso kein Markt für die Hersteller. Eine Abgabe zu Herstellungskosten würde also keinen Verlust für die Firmen darstellen. Zweitens steht die Rechtfertigung an sich auf tönernen Füßen: Die WHO hat die Forschungsausgaben für 99 Krebsmedikamente, die zwischen 1989 und 2017 in den USA auf den Markt kamen, mit den erzielten Umsätzen für diese Mittel verglichen. Dabei kamen sie auf einen durchschnittlichen Umsatz von 14,50 US$ für jeden in die Forschung investierten Dollar (einschließlich Kosten für Fehlschläge).[9] Krebsmedikamente sind also ein äußerst lukratives Geschäft. Leider eines das auf Kosten der Armen dieser Welt geht.
Gesundheitsversorgung für Alle ist ein erklärtes Ziel der Vereinten Nationen. Dazu gehört eine rationale Auswahl der verwendeten Medikamente ebenso wie die Senkung der Preise auf ein bezahlbares Maß. (JS)
Artikel aus dem Pharma-Brief 4-5/2019, S.1
Bild Pillen © ironstealth /istock
[1] Centre for Urban Health Solutions, University of Toronto und Centre for Evidence-Based Medicine, University of Oxford
[2] Persaud N et al. (2019) Comparison of essential medicines lists in 137 countries. Bull WHO; 97, p 394
[3] WHO (2017) 20th Essential Medicines List. www.who.int/medicines/news/2017/20th_essential_med-list/en
[4] Bei etlichen Wirkstoffen sieht die WHO eine Äquivalenz innerhalb der Wirkstoffgruppe (in der WHO-Liste mit einem Quadrat markiert). Bei diesen Wirkstoffen wurde in der vorliegenden Untersuchung die Gleichwertigkeit angenommen und die nationale Auswahl wurde in diesem Fall nicht als Abweichung von der WHO-Liste gewertet.
[5] https://global.essentialmeds.org [Zugriff 12.7.2019]
[6] arznei-telegramm (2017) Arzneimitteldatenbank [Zugriff 12.7.2019]
[7] ‚t Hoen E et al. (2019) Improving affordability of new Essential Cancer Medicines. Lancet Oncol http://dx.doi.org/10.1016/S1470-2045(19)30459-0
[8] WHO (2019) 21th Essential Medicines List. https://apps.who.int/iris/handle/10665/325771
[9] WHO (2018) Pricing of cancer medicines and its impacts. Geneva: WHO https://apps.who.int/iris/bitstream/handle/10665/277190/9789241515115-eng.pdf
Die schmutzige Rückseite der Medaille
UN verletzt eigene Prinzipien durch fragwürdige Kooperationen
Public-Private-Partnerships[1] (PPPs) zwischen staatlichen und privaten Akteuren finanzieren weltweit zahlreiche (semi-)öffentliche Projekte. Ein kürzlich erschienenes Arbeitspapier von MISEREOR, Brot für die Welt und Global Policy Forum wirft einen kritischen Blick auf die Partnerschaften der UN.[2]
PPPs sind nicht nur auf nationaler Ebene im Trend, um das Problem knapper öffentlicher Gelder zu umgehen. Auch die Vereinten Nationen (UN) setzen bei der Umsetzung ihrer Ziele für nachhaltige Entwicklung (Sustainable Development Goals, SDGs) vermehrt auf die Zusammenarbeit mit privaten Unternehmen. Die zunehmende Verstrickung der Wirtschaft in Projekte und Politik der UN birgt jedoch einige Risiken. Wirtschaftsakteure verfügen im Vergleich zu zivilgesellschaftlichen Akteuren ohnehin bereits über einen besseren Zugang zu den Entscheidungsprozessen in der UN. Durch die PPPs und die damit einhergehenden stärker formalisierten Beziehungen droht sich das Ungleichgewicht zwischen wirtschaftlichen und zivilgesellschaftlichen Interessen in der globalen Politik noch mehr auszuweiten.
Gleichzeitig gibt es mehrere Fälle von sogenanntem Bluewashing, also dem Schönfärben des Unternehmens durch eine Kooperation mit der UN. Dabei soll die Assoziation mit der UN und der vermeintliche Einsatz für die SDGs das Image des Unternehmens aufbessern, obwohl die tatsächliche Unternehmenspraxis nicht mit den Nachhaltigkeitszielen vereinbar ist.
Ein Beispiel dafür ist die Partnerschaft zwischen der Bill & Melinda Gates Stiftung und der WHO. Die philanthropische Stiftung investiert gleichzeitig in Unternehmen der Nahrungsmittel- und Getränkeindustrie, wie Coca-Cola, Walmart und dem weltweit größten Franchiseträger von McDonalds. Die finanziellen Bindungen zu diesen Konzernen stehen im völligen Gegensatz zu den Werten der WHO und den Nachhaltigkeitszielen der UN. Hieran wird deutlich, dass die Interaktion zwischen UN und Wirtschaft nicht ausreichend geregelt ist. Daher fordern die AutorInnen strengere, aber vor allem allgemein gültige und angewendete Regularien, anhand derer potentielle Partnerschaften von UN-Institutionen mit Akteuren aus dem privaten Sektor gemessen werden sollen. (MB)
Artikel aus dem Pharma-Brief 10/2019, S. 6
[1] Public-Private-Partnership ist zwar in diesem Kontext der übliche Begriff, dennoch erachten wir ihn als unpassend, da die Einordnung als Partnerschaft eine Beziehung auf Augenhöhe und mit gemeinsames Zielen suggeriert. In der Realität stehen die öffentlichen und unternehmerischen Ziele häufig im Kontrast zueinander.
[2] Martens J and Seitz K (2019) Rules of engagement between the UN and private actors. Towards a regulatory and institutional framework. p. 10. www.globalpolicy.org/images/pdfs/Rules_of_Engagement_UN_Private_Actors_web.pdf [Zugriff 10.10.2019]
Deutsche Kurzfasssung: Seitz K (2019) Nachhaltig nur auf dem Papier? Die ambivalente Rolle der Wirtschaft bei der Umsetzung der SDGs. S.3. www.globalpolicy.org/images/pdfs/Briefing_0319_Nachhaltig_nur_auf_dem_Papier.pdf [Zugriff 10.10.2019]
Die nächste Schlappe
Brustkrebsmedikament bleibt ohne Zusatznutzen
Das Krebsmedikament Palbociclib war mit viel Vorschusslorbeeren gestartet. Doch nun kommt der nächste Dämpfer. Der Gemeinsame Bundesausschuss entschied nach gründlicher Auswertung einer zweiten Studie endgültig, dass es keinen zusätzlichen Nutzen gibt.
Zur Erinnerung: Mit drei Studien versucht(e) der Hersteller die Vorteile von Palbociclib[1] zu belegen: Mit Paloma 1 und 2 für Frauen mit Brustkrebs in der Erstlinientherapie[2] und mit Paloma 3 für Frauen nach Vortherapien.[3] Bei der Zulassung war keine der drei Studien abgeschlossen und Vorteile für das Überleben waren noch nicht belegt.[4] Die erste Nutzenbewertung vom Mai 2017 durch den Gemeinsamen Bundesausschuss (G-BA) [5] verlief für alle Patientinnengruppen negativ, das Urteil lautete „kein Zusatznutzen“ für Palbociclib. Da zum Zeitpunkt der Beschlussfassung alle Studien noch liefen bzw. keine endgültigen Ergebnisse vorlagen, war der Beschluss befristet.[6]
Bereits direkt nach diesem Beschluss wurden die Endergebnisse von Paloma 1 bekannt: Kein Überlebensvorteil für die betroffenen Frauen – aber mehr Nebenwirkungen.[4] Im Sommer 2018 wurden die Endergebnisse von Paloma 3 für die Behandlung von Frauen, die bereits Vortherapien erhalten hatten, bekannt: In dieser Studie sicherte Palbociclib ebenfalls kein längeres Überleben. Obwohl der Hersteller versuchte, die Resultate schönzurechnen,[7] blieb der G-BA jetzt in einer neuen Bewertung bei seinem ursprünglichen Urteil, dass für diese Patientinnen kein Zusatznutzen durch den neuen Wirkstoff belegt ist.[8]
Das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) hatte dafür die Daten des Herstellers genau unter die Lupe genommen und der G-BA sich in der anschließenden Diskussion große Mühe gegeben. Der G-BA hob mehrere Kritikpunkte hervor. So wurden nachträglich mehr Frauen in die Studie aufgenommen als ursprünglich geplant – und das, wo für den Hersteller nicht so erfreuliche Zwischenergebnisse schon bekannt waren. Das riecht nach Manipulation. Mindestens ebenso wichtig: Ungewöhnlich viele Frauen waren während der Paloma 3-Studie „verlorengegangen“, für sie lagen keine Daten zum Überleben vor. Dass die für diese Patientinnengruppe gewählte Vergleichstherapie Fulvestrant als alles andere als optimal gilt, ist ein weiterer Schwachpunkt.
Nachdem nun für drei von vier Patientinnengruppen das Urteil „kein Zusatznutzen“ feststeht, bleibt als letzter Rettungsanker für den Hersteller für die Erstlinientherapie bei Frauen nach der Menopause nur noch die Paloma 2-Studie. Bisherige Zwischenergebnisse zeigen aber (noch) keinen Überlebensvorteil. Und die Dauer der Studie wurde schon zweimal verlängert, um doch noch positive Ergebnisse zu erzielen. Die erste Frist zur Vorlegung der Daten beim G-BA lief bis zum 1.3.2019, aber der Hersteller signalisierte schon letztes Jahr, dass er sie nicht einhalten kann. Jetzt müssen die Daten bis zum 2.1.2021 vorgelegt werden.
Man rufe sich in Erinnerung: Ende 2016 wurde Palbociclib aufgrund wenig tragfähiger Daten zugelassen. Erst vier Jahre danach kann eine aussagekräftige Bewertung des Nutzens für eine nicht unwesentliche Gruppe von PatientInnen begonnen werden. Klarheit, ob wenigstens eine Patientinnengruppe von dem Medikament profitiert, gibt es also erst Mitte 2021 nach Abschluss der Bewertung durch den G-BA.
Wem nützt es?
Der Wirkstoff kostet über 66.000 € pro Patientin und wird zusätzlich zur bisherigen Medikation gegeben. Für den Hersteller ein gutes Geschäft.
Für Frauen, die von Brustkrebs betroffen sind, sind die Ergebnisse der unabhängigen Bewertung von Palbociclib enttäuschend. Denn sie hoffen zu Recht auf bessere Behandlungsmöglichkeiten. Allerdings darf man dabei nicht aus den Augen verlieren, dass der Hersteller den Hype um das Medikament kräftig geschürt hat. Leider machen leere Versprechen niemand gesund. (JS)
Artikel aus dem Pharma-Brief 2/2019, S. 6
[1] Palbociclib ist zugelassen für Hormonrezeptor (HR)-positiven, humanen epidermalen Wachstumsfaktor-Rezeptor-2(HER2)-negativen lokal fortgeschrittenen oder metastasierten Brustkrebs
[2] Zur Erstlinientherapie in Kombination mit einem Aromatasehemmer
[3] In Kombination mit Fulvestrant bei Frauen, die zuvor eine endokrine Therapie erhielten
[4] Pharma-Brief (2017) Viel Lärm um nichts? Nr. 4, S. 4
[5] G-BA Beschluss vom 18.5.2017 www.g-ba.de/bewertungsverfahren/nutzenbewertung/269/#tab/beschluesse
[6] Ausnahme: Erstlinientherapie bei Frauen vor der Menopause. Dazu hatte der Hersteller keine Studie durchgeführt. Für diese Patientinnengruppe war der Beschluss vom 18.5.2017 endgültig.
[7] Pharma-Brief (2018) Brustkrebs: Leere Versprechen. Nr. 6, S. 4
[8] G-BA (201) Beschluss vom 22.3.2019 www.g-ba.de/bewertungsverfahren/nutzenbewertung/394/#tab/beschluesse
Die Kultur verändern
Bessere Kommunikation verbessert Antibiotika-Verordnungen
2016 gründeten Kinder- und Jugendärzte in Bielefeld das Projekt AnTiB (Antibiotische Therapie in Bielefeld). Ihr Ziel: Auf lokaler Ebene für den ambulanten Bereich einheitliche, praxistaugliche und möglichst breit akzeptierte Regeln zur Verschreibung von Antibiotika zu entwickeln. Roland Tillmann, Kinderarzt und Mitbegründer der Initiative gibt Auskunft über die selbstgesteckten Ziele, erste Erfolge und Zukunftsperspektiven.
Herr Tillmann, gab es einen konkreten Anstoß für die Gründung von AnTiB?
Im Verschreibungsverhalten von ÄrztInnen gibt es sehr große Unterschiede. Das führt im Arbeitsalltag zu Konflikten – mit den Patienten bzw. mit besorgten Eltern, denen man die unterschiedliche Herangehensweise erklären muss, aber auch mit den Kollegen anderer Fachrichtungen oder aus anderen Sektoren. Jemand, der Antibiotika zurückhaltend und nur gezielt verordnet, sieht sich immer wieder solchen Konflikten ausgesetzt. Da gab es also fachlichen und persönlichen Leidensdruck.
Andererseits existieren in Bielefeld schon länger gute Kommunikationsstrukturen – etwa mit der Initiative Bielefelder Hausärzte und der Kinderklinik Bethel. Was fehlte war eine gemeinsame Kommunikationsstrategie. Aber dieses Potenzial konnte man nutzen, um das Verschreibungsverhalten besser abzustimmen. Das haben wir getan und Anfang 2017 erstmals Empfehlungen zur Antibiotika-Verordnung in der ambulanten pädiatrischen Versorgung erarbeitet und veröffentlicht.
Wie genau geschah das?
Niedergelassene Kinderärzte und Vertreter der örtlichen Kinderklinik haben das Projekt gestartet. In einem mehrstufigen, moderierten Kommunikationsprozess haben wir uns auf Empfehlungen zur Standardbehandlung häufiger Infektionskrankheiten verständigt. Am Ende stand ein breiter Konsens über eine einheitliche Verordnungspraxis – kurz gefasst, anwendungs- und anwenderorientiert. Immer wieder wurden Beiträge per E-Mail herumgeschickt, zur Diskussion gestellt und Kommentare von Kollegen eingearbeitet. Um eine externe Qualitätssicherung zu garantieren, wurden zudem Experten der Deutschen Gesellschaft für pädiatrische Infektiologie (DGPI) in den Prozess einbezogen.
Was war das erklärte Ziel?
Konkretes Ziel war, die Schnittstellenproblematik zu verbessern. Wenn eine Mitbehandlung z.B. im Notdienst oder in der Klinik stattfindet, sollte es eine bessere Zusammenarbeit und eine einheitliche Gesprächsführung mit dem Patienten geben. Das haben wir erreicht.
Den Antibiotikaverbrauch zu reduzieren, war zunächst gar nicht so sehr unsere Intention gewesen. Diese Zielsetzung rückte erst durch die Deutsche Antibiotika-Resistenzstrategie (DART)[1] der Bundesregierung in den Fokus. Damit hat das Projekt einen neuen Charakter bekommen. DART schlägt ja als Maßnahme u.a. die Erarbeitung von Konzepten zur Erstellung und Anwendung lokaler Leitlinien und Empfehlungen im ambulanten und stationären Bereich vor. Lokale Antibiotika-Leitlinien für die ambulante Medizin gab es aber vorher nicht. Andere Projekte wurden von universitären Einrichtungen angestoßen und legen den Schwerpunkt auf externe Schulungen. Die entscheidende Besonderheit von „AnTiB“ ist, dass das Projekt von der Basis ausging, also eine Initiative von praktizierenden Ärztinnen und Ärzten ist.
30 bis 50 Prozent aller Antibiotika werden laut Schätzungen ohne medizinische Notwendigkeit verschrieben. Warum ist es trotz zahlreicher Leitlinien und Fortbildungen so schwer, das Verschreibungsverhalten zu verändern?
Antibiotika-Verordnungen finden auch in einem sozialen Kontext statt. Da spielen z.B. die Macht von Erfahrungen und Gewohnheiten und auch vermutete Erwartungshaltungen eine Rolle und Entscheidungen unter Zeitdruck.
… wie kann AnTiB darauf Einfluss nehmen?
Indem wir Verordnungen nicht nur als individuelles Verhalten sehen, sondern auch als ein kulturelles Phänomen. Verordnungen werden nicht automatisch besser, wenn wir nur mehr infektiologisches Wissen verbreiten. Wir müssen vielmehr Normen bzw. die Kultur verändern, die den Entscheidungen auch zugrunde liegen. Eine Studie spricht hier von „local cultural unspoken rules“, also verdeckten kulturellen Regeln. In Nordrhein-Westfalen werden z.B. fast 50 Prozent mehr Antibiotika verschrieben als in den östlichen Bundesländern. Und auch an den Ländergrenzen – etwa zu den Niederlanden – ändert sich das Verschreibungsverhalten. Es geht also um kulturelle Unterschiede.
Genau hier setzt AnTiB an: Denn die lokale Ebene ist der Ort, wo wir den sozialen Kontext verändern und neue Normen prägen können, indem wir uns austauschen, miteinander Absprachen treffen.
Warum spielt dabei gerade der ambulante Bereich eine große Rolle?
85 Prozent aller humanmedizinischen Antibiotika-Verordnungen entfallen auf ambulante Arztpraxen. Ansätze zum rationaleren Antibiotika-Gebrauch beziehen sich aber bisher eher auf die Krankenhäuser. Im ambulanten Bereich besteht ein großer Bedarf an innovativen Konzepten.
Zeigen sich bereits erste Erfolge von AnTiB?
AnTiB zieht Kreise. Das Konzept wurde lokal auf andere Fachrichtungen ausgeweitet – Hausärzte und Gynäkologen, Urologen und HNO-Ärzte, weitere Fachrichtungen sollen folgen. Wir beteiligen uns außerdem an überregionalen Absprachen und haben unser Bielefelder Konzept in vielen anderen Städten und bei Fachkongressen vorgestellt.
Konflikte treten jetzt im Praxisalltag kaum mehr auf. Inzwischen ist es Standard, die Patienten im Zweifelsfall noch einmal zur Kontrolle zu bestellen, statt „vorsorglich“ ein Antibiotikum zu verschreiben und das auch so zu kommunizieren. In Umfragen zeigte sich bei allen Kollegen eine große Zufriedenheit. Selbst von denen, die anfänglich eher skeptisch waren, kamen sehr positive Rückmeldungen.
Inwieweit auch die Antibiotika-Verordnungen reduziert wurden, wird bald eine wissenschaftliche Evaluation des Projektes durch die Fakultät für Gesundheitswissenschaften der Universität Bielefeld zeigen.[2]
Ihre ganz persönliche Erkenntnis aus dem Projekt?
In der Medizin brauchen wir viel mehr Kooperationsfähigkeit und Kooperationsbereitschaft. Dies ist nicht nur eine Voraussetzung für Projekte wie AnTiB, sie steigern genau solche Fähigkeiten auch.
Konzeption und Materialien von AnTiB werden Ärztinnen und Ärzten auch überregional zur Anwendung zur Verfügung gestellt. Weitere Informationen: www.antib.de, Kontakt: Diese E-Mail-Adresse ist vor Spambots geschützt! Zur Anzeige muss JavaScript eingeschaltet sein!
Foto Roland Tillmann © Susanne Freitag
Artikel aus dem Pharma-Brief 6/2019, S.6
[1] Informationen zu DART: www.bundesgesundheitsministerium.de/themen/praevention/antibiotika-resistenzen/antibiotika-resistenzstrategie.html
[2] Informationen zu AnTiB und der wissenschaftlichen Projektbegleitung durch die Fakultät für Gesundheitswissenschaften der Uni Bielefeld: www.antib.de
Die Geister von Genf
Transparenz bei Forschung weiter auf internationaler Agenda
Nach der viel diskutierten Kontroverse um Transparenz bei Arzneimittelpreisen auf der Weltgesundheitsversammlung im Mai in Genf hält die Debatte an. Im Vorfeld der nächsten UN Vollversammlung zeigen sich abermals alte Konfliktlinien. Welche Rolle wird Deutschland einnehmen?
„Nach dem Spiel ist vor dem Spiel“ – diese Sportlerweisheit gilt momentan auch in der Debatte um verbesserten Zugang zu medizinischer Behandlung. Zwar endete die letzte Weltgesundheitsversammlung in Genf beim Thema Transparenz-Resolution mit einem Paukenschlag und scharfer deutscher Kritik (wir berichteten[1]). Doch nehmen nachfolgende Prozesse den Faden direkt auf
Am 23. September findet in New York im Zuge der UN Vollversammlung das UN High-Level Meeting zu Universal Health Coverage (UHC) statt. Im sehr umfangreichen UHC-Resolutionsentwurf finden sich auch Aussagen zu Transparenz und Forschung. Ein Umstand, der angesichts der zurückliegenden Ereignisse in Genf gerade aus deutscher Perspektive beachtenswert ist.
Ein hartnäckiges Thema
Bis heute zieht sich die Bundesregierung auf den schwachen Einwand zurück, dass das damalige Prozedere bei der Transparenz-Resolution fehlerhaft gewesen sei und man sich deshalb distanziert habe.[2] Eine echte Auseinandersetzung mit der Tatsache, dass sich Deutschland vorher massiv für die Verwässerung und Beschneidung der Inhalte ins Zeug gelegt hatte, fand zumindest öffentlich nicht statt. Nun bildeten jedoch abermalige Debatten um Transparenz und Forschung eine der größten Hürden bei der Vorbereitung des Textes für New York.[3]
Gerungen wurde um mehrere Punkte, ein wichtiger war das Plädoyer für die Erhöhung der Transparenz bei Preisen und Forschungskosten. Hier hatte es bereits bei der Weltgesundheitsversammlung massive Konfrontationen zwischen Mitgliedsstaaten gegeben. Weitere umstrittene Ausführungen setzten sich für verbesserte Förderung alternativer Produktentwicklung ein. Zentral ist hier das Streben, den Produktpreis von den Kosten für Forschung und Entwicklung zu entkoppeln. „De-Linkage“, der verbreitete Begriff für diesen Ansatz, wurde jedoch offenbar absichtlich ausgespart, um Anstoß zu vermeiden. Ein später Entwurf enthielt zudem die Analyse, dass hohe Preise und schlechter Zugang Fortschritte bei UHC anhaltend beeinträchtigen. Auch diese Textstelle blieb umkämpft.[3]
New York ist nicht Genf – oder?
Universal Health Coverage ist ein vielschichtiges Thema, bei dem die Aspekte Transparenz und Forschung nur zwei von vielen sind. Allerdings können sie große Auswirkungen auf den Zugang zu angemessener Gesundheitsversorgung haben, gerade in ärmeren Ländern. So rechnet die WHO damit, dass allein in Afrika jedes Jahr elf Millionen Menschen durch Gesundheitsausgaben in die Armut rutschen.[4] Dabei geht es beispielsweise längst nicht nur um vernachlässigte Infektionskrankheiten, sondern auch verstärkt um nicht-übertragbare Krankheiten. Deutlich sichtbar ist dies etwa bei hochpreisigen Krebsmedikamenten.[5]
Die Bundesregierung wird im September voraussichtlich hochrangig in New York vertreten sein. In Genf hatte sich gezeigt, dass die öffentliche Aufmerksamkeit für die Themen Transparenz und Zugang groß ist und nationale Positionen kritisch, sowie mit Nachdruck hinterfragt werden. Es ist zu hoffen, dass die deutsche Politik daraus Schlüsse gezogen hat und diesmal eine konstruktivere Haltung einnehmen wird. (MK)
Artikel aus dem Pharma-Brief 4-5/2019, S.3
[1] Pharma Brief (2019) WHA: Deutschland auf Distanz zu Transparenz-Beschluss. Nr. 3. S. 1
[2] Dabei ist das Fundament dieser Kritik sehr fraglich. Denn Resolutionsentwürfe können nach WHO-Regularien bis zum ersten Tag der Weltgesundheitsversammlung eingebracht werden, wenn die Resolution auf der Agenda ist. Dies war bei der Transparenz-Resolution der Fall. Es ist zudem nirgendwo vorgegeben, dass Resolutionen zunächst beim Board eingebracht werden müssen, was ebenfalls ein deutscher Beschwerdepunkt war.
[3] Health Policy Watch (2019) Drug R&D, Sexual & Reproductive Health Scrutinised In Draft UHC Declaration. www.healthpolicy-watch.org/drug-rd-sexual-reproductive-health-scrutinised-in-draft-uhc-declaration/ [Zugriff 23.07.2019]
[4] DEVEX (2019) Achieving UHC in Africa requires support for most vulnerable, experts say www.devex.com/news/achieving-uhc-in-africa-requires-support-for-most-vulnerable-experts-say-94411 [Zugriff 24.07.2019]
[5] 't Hoen E et al. (2019) Improving affordability of new Essential Cancer Medicines. Lancet Oncol http://dx.doi.org/10.1016/S1470-2045(19)30459-0
Das Beste draus machen
Wie man negative Studienergebnisse kleinredet
„ ‘Alternative Fakten‘ zur Erklärung enttäuschend negativer Studienergebnisse?“ titelte Arzneimittelbrief.[1] Basis des Artikels war eine kleine Studie, die in der Weihnachtsausgabe des BMJ erschienen war.[2] Mit einer guten Prise britischen Humors gewürzt, beleuchtete sie das professionelle Schönreden von wissenschaftlichen Niederlagen.
Schon die Einleitung des BMJ-Artikels hat es in sich: „Der Fortschritt in der medizinischen Wissenschaft wird getrübt durch häufiges auf der Stelle treten, weil die meisten wundervollen Ideen am Ende nicht funktionieren. Um den Eindruck einer Flut von Entdeckungen und Begeisterung aufrechtzuerhalten, heuern Pharma- und Medizinproduktehersteller selektiv ExpertInnen ihres Fachs an, bei denen sie sich in allen Situationen auf eine ermutigende Darstellung verlassen können. Intern nennen die Firmen sie ‚Key Opinion Leader‘. Wir haben festgestellt: Wenn Key Opinion Leader enttäuschende Studienergebnisse in Nachrichtenportalen oder auf Konferenzen kommentieren sollen, scheinen sie merkwürdig unfähig zu erkennen, dass die Behandlung nicht funktioniert.“ Sie (er)finden zahlreiche Gründe, warum das Medikament doch besser sein könnte, als es die Studie ergab.
Da die sechs AutorInnen den Eindruck hatten, dass sich die Ausreden für die schlechten Ergebnisse häufig wiederholten, gingen sie die Sache systematisch an. Sie werteten Kommentare zu neuen Studien aus, die 2013-2017 auf drei großen Kardiolog-Innenkongressen in den USA und Europa vorgestellt worden waren. Die ForscherInnen fanden in den vielgelesenen Nachrichtenportalen „Medscape“ und „MedPage Today“ Kommentare zu 127 für den Sponsor unvorteilhaft ausgegangenen Studien.[3] In den allermeisten Fällen wurden die enttäuschenden Ergebnisse in Frage gestellt: In 108 der Berichte fanden sich Rechtfertigungen, warum das negative Ergebnis nicht aussagekräftig sei.
Die beliebteste Ausrede war, dass angeblich zu wenig PatientInnen in die Studie eingeschlossen waren, um ein statistisch signifikantes Ergebnis zu erzielen. Nur in einem von 39 Fällen machte sich der Key Opinion Leader aber die Mühe, die notwendige Anzahl vorzurechnen.
Die Ausrede „es sind weitere Studien notwendig“ (jede fünfte Negativstudie wurde damit kleingeredet) kommentieren die AutorInnen so: „Das legt nahe, dass der Key Opinion Leader die Ergebnisse einfach nicht mochte und wünscht, dass die Würfel erneut geworfen werden.“
Auch beliebt war die Behauptung, dass die PatientInnen zu krank waren (7 mal), um von der Behandlung noch profitieren zu können. Allerdings wurde bei elf Studien das negative Ergebnis mit dem genauen Gegenteil erklärt, dass die Krankheit zu mild oder in einem zu frühen Stadium war. Mal waren die Patientinnen nach dem Urteil der Propagandisten zu jung, mal zu alt, die Compliance war zu gut oder zu schlecht.
Mit typisch englischem Humor verfassten die AutorInnen am Schluss ein Drehbuch für die Key Opinion Leader. Ihr Motto: „Mit Hilfe des Drehbuchs ist keine Intervention zu ineffektiv, als dass sich nicht eine Ausrede dafür finden ließe.“ (JS)
Artikel aus dem Pharma-Brief 1/2019, S.5
[1] Der Arzneimittelbrief (2019) 53; S. 16DB01
[2] Hartley A et al. (2018) Key opinion leaders‘ guide to spinning a disappointing clinical trial result. BMJ; 363, p k5207
[3] Für den primären Endpunkt der Studie konnte kein signifikanter Unterschied zur Vergleichsbehandlung festgestellt werden.
Außer Kontrolle
Ebola-Epidemie im Kongo spitzt sich weiter zu
Ein Jahr nach dem Ausbruch einer Ebola-Epidemie im Osten der Demokratischen Republik Kongo ist die Seuche noch immer auf dem Vormarsch. In den betroffenen Gebieten steigt die Rate der Neuinfektionen – trotz massiver Bekämpfungsmaßnahmen. Wir sprachen mit dem Tropenmediziner Maximilian Gertler von Ärzte ohne Grenzen über Behandlungsprogramme, Präventionsarbeit und die Hintergründe der gegenwärtigen Lage.
Die WHO hat erst kürzlich kundgetan, dass es sich im Kongo nicht um einen internationalen Gesundheitsnotstand handelt. Wie beurteilen Sie die Lage vor Ort?
Dass kein internationaler Gesundheitsnotstand erklärt worden ist, finde ich zwar nachvollziehbar, weil die Epidemie bisher weitgehend auf eine Region begrenzt ist. Aber dass der Ausbruch nicht unter Kontrolle ist, ist offensichtlich. Er hat in absoluten Zahlen Ausmaße erreicht, die man bisher nur von der großen Epidemie in Westafrika vor 5 Jahren kannte: Über 2.000 Infizierte und über 1.500 Todesopfer seit Juni 2018. Man kriegt die Krankheit momentan nicht in den Griff – vor allem wegen der Konfliktsituation. Immer wieder gab es Angriffe auf Behandlungszentren und Helfer, z.B. in der Stadt Butembo.
Was sind die Gründe für diese Attacken?
Innerhalb der Bevölkerung herrscht ein großes Misstrauen gegenüber den Hilfsmaßnahmen. Eine im März im Lancet veröffentlichte Untersuchung hat gezeigt, dass über die Hälfte der Befragten glaubt, Ebola existiere nicht oder sei erfunden worden, um die Region zu destabilisieren. Es kursieren wilde Gerüchte über die Bekämpfungsmaßnahmen. Eine im März 2019 veröffentlichte Befragung von Translators without Borders zeigt, dass Poster, Broschüren und anderes Informationsmaterial vielfach nicht oder falsch verstanden werden. Vertrauen ist aber entscheidend für eine erfolgreiche Prävention und Prävention ist der Schlüssel zur Beendigung der Epidemie.
Woher rührt das mangelnde Vertrauen?
Die Bevölkerung fühlt sich vernachlässigt. Sie empfindet die staatliche Sorge um Ebola angesichts des jahrzehntelangen Konflikts als unglaubwürdig. Seit Jahren erfahren die Menschen unbeschreibbare Gewalt: Die ständige Angst vor Vertreibung, Überfällen, Vergewaltigung. Kinder, die für Söldnergruppen rekrutiert werden… Es sterben wegen der mangelhaften medizinischen Versorgung sehr viele Menschen auch an anderen Krankheiten. Neben all diesem Leid hat Ebola für die Menschen im Ost-Kongo eine untergeordnete Bedeutung.
Gegen Ebola gibt es inzwischen einen Impfstoff, der in Kanada mit öffentlichen Geldern erforscht, an ein kleines Pharmaunternehmen auslizensiert und 2014 während des Ebola-Ausbruchs in Westafrika von der US-Firma Merck aufgekauft wurde. Noch immer ist das Mittel nicht zugelassen und wird unter Studienbedingungen eingesetzt. Wie gut ist dieser Impfstoff verfügbar?
Nur eine Woche, nachdem der Ebola-Ausbruch im Kongo bestätigt wurde, stand der Impfstoff zur Verfügung. Er ist offenbar sehr gut wirksam und gut verträglich. Ärzte ohne Grenzen hat Impfteams ausgebildet und in die Regionen geschickt. Im Rahmen von Ring-Impfkampagnen wurden zunächst alle Gesundheitshelfer, dann alle Kontaktpersonen von Infizierten und schließlich die Kontakte der Kontaktpersonen geimpft. Bis jetzt wurden mehr als 136.000 Menschen im Ost-Kongo unter Führung der WHO und des nationalen Gesundheitsministeriums geimpft. Ich kann nicht sagen, wieviel Impfstoff in der Region noch verfügbar ist. Merck hat weitere Lieferungen angekündigt, und gegenwärtig erklärt die WHO die Möglichkeit, mit reduzierten Impfdosen zu arbeiten.
Was geschieht, wenn der Impfstoff zur Neige geht?
Die WHO hat erklärt, dass die Impfstoffdosis bei Kontaktpersonen auf die Hälfte und bei den Kontaktpersonen der Kontaktpersonen auf ein Fünftel reduziert werden kann, um so mehr Menschen mit dem vorhandenen Vorrat impfen zu können. Das kongolesische Gesundheitsministerium ist außerdem dabei, weitere Optionen zu prüfen – gemeinsam mit der WHO, der London School of Hygiene & Tropical Medicine und uns. Dabei geht es zum Beispiel um den möglichen Einsatz eines zweiten Impfstoffs von Johnson & Johnson, der sich derzeit in klinischen Studien der Phase 3 befindet. Von diesem Impfstoff sollen in absehbarer Zeit 1,5 Millionen Impfdosen zur Verfügung stehen. Damit könnten dann nicht mehr nur unmittelbar betroffene Personen, sondern ganze Regionen durchgeimpft werden.
Aufgrund der Angriffe musste Ärzte ohne Grenzen seine Behandlungszentren im Ost-Kongo schließen. Wie geht es jetzt weiter?
In Butembo und Katwa, den noch vor Wochen am stärksten betroffenen Gebieten, sind die Strukturen, die wir hatten, zerstört. Isolierstationen betreiben wir nur noch in anderen Regionen. Aber wir unterstützen weiterhin vor allem die epidemiologische Arbeit, Impfmaßnahmen und Aufklärungskampagnen. Unser Fokus liegt darauf, die vorhandenen Gesundheitsstrukturen im Kampf gegen Ebola zu stärken. Man muss den Menschen dort helfen, wo sie sind. Schließlich ereignet sich etwa die Hälfte der Todesfälle in den Dörfern und Familien und nicht auf den Isolierstationen. Und es geht nicht nur um Ebola – es geht um die Gesundheit der Bevölkerung insgesamt.
Artikel aus dem Pharma-Brief 3/2019, S.3
Antibiotikaresistenzen in NRW-Gewässern
BUND fordert Konsequenzen
Die Naturschutzorganisation BUND hat in Flüssen und Seen in NRW nach resistenten Keimen gesucht – und wurde fündig. Paul Kröfges, Gewässerexperte des BUND NRW, berichtet.
Nachdem der NDR Anfang 2018 antibiotikaresistente Bakterien (ARB) in mehreren Gewässern Niedersachsens nachgewiesen hatte, stellte sich die Frage nach der Situation in anderen Bundesländern. In Nordrhein-Westfalen wiegelte das zuständige Umweltministerium ab und verwies auf ein länger laufendes Forschungsprojekt (HyReKA),[1] dessen Ergebnisse man abwarten wolle.
Daraufhin beauftragte der BUND NRW im April und Juli 2018 die Medizinische Fakultät der Ruhr Universität Bochum mit der Untersuchung von 13 Gewässerproben aus NRW nach einem anerkannten Verfahren.[2] Es handelte sich dabei um 5 Proben aus landwirtschaftlich geprägten Gewässerstrecken im Kreis Borken, einer Probe aus einem Badesee bei Viersen und weiteren 7 Proben aus Gewässern in NRW, die unterschiedliche Einflüsse aus Kläranlagen aufweisen.[3]
Untersuchungen 2018
In den landwirtschaftlich beeinflussten Proben wurden bis zu 7 Resistenzen gegenüber 14 getesteten Antibiotika festgestellt. Dies sind deutliche Hinweise auf den Einsatz von Antibiotika in der dort praktizierten Tierhaltung und der Resistenzverbreitung über die Ausbringung belasteter Gülle.
In der Probe aus einem Badesee bei Viersen wurden Colibakterien mit medizinisch relevanter Multiresistenz gegen 3 Reserveantibiotika nachgewiesen (3 MRGN). Als Ursachen kommen sowohl Einträge durch Zuflüsse von landwirtschaftlichen Flächen als auch durch Badende in Frage.
Problematisch sind auch die Ergebnisse aus Nette, Sieg und Agger. So wurden in einem Abschnitt der Nette, vor der Kläranlage Viersen-Dülken, hohe Anteile an multiresistenten Colibakterien gefunden (3 MRGN). Ursache dafür könnte der Überlauf von Mischwasser aus dem überlasteten Kanalnetz [4] oder auch eine nahegelegene große Schweinemastanlage sein.
Auffallend war die Belastung hinter Kläranlagen mit angeschlossenen Krankenhäusern. So lagen hinter der Kläranlage Rosbach (ohne Klinik) in der Sieg nur relativ geringe Belastungen vor. Dagegen wurden weiter flussabwärts, etwa 2 km hinter der Kläranlage Eitorf, die Abwässer eines Krankenhauses aufnimmt, resistente Bakterien der Kategorie 3 MRGN festgestellt. Zwischen dem Ablauf der Kläranlage und der Probenentnahmestelle liegt ein Campingplatz mit großem Badebetrieb bei sommerlichen Temperaturen.
Als noch problematischer erwies sich die Situation an der Agger, einem Nebengewässer der Sieg. Dort wurden hinter der Kläranlage Engelskirchen, an die 2 größere Kliniken angeschlossen sind, in einer Probe vom 10. Juli 2018 sogar 4 MRGN (E-Coli) Bakterien nachgewiesen.
Hier ist die Situation des Gewässers besonders problematisch, da die Einleitung der Kläranlage in das alte Aggerbett bei Ehreshoven erfolgt. Das Flüsschen hat eine Mindestwasserführung von nur 500 Litern Wasser pro Sekunde, während der weitaus größere Wasseranteil nebenan kanalisiert in die Turbinen der Aggerkraftwerke geleitet wird. Der Rest im ehemaligen natürlichen Flussbett muss mit dem keim- und spurenstoffbelasteten Abwasserstrom fertig werden. Unter diesen Bedingungen können sich unterhalb des Kläranlagenablaufes antibiotikaresistente Bakterien vermehren, u.U. sogar regelrecht gezüchtet werden, so die Befürchtung des BUND.
Untersuchungen 2019
Da der zuständige Wasserverband keine aktuellen Messwerte von Arzneimitteln, insbesondere Antibiotika, vorlegen konnte, wurde der BUND erneut aktiv. Er ließ im März und April 2019 zwei Proben aus dem Ablauf der Kläranlage Engelskirchen auf 42 Komponenten aus diesem Spektrum untersuchen.
Die Ergebnisse belegen nach Einschätzung des BUND eine deutliche Belastung der Agger mit Rückständen von Antibiotika, Blutdrucksenkern, Epilepsie-, Herz- und Kreislaufmitteln verschiedenster Art durch die Kläranlage Engelskirchen.
Konsequenzen und Forderungen
Der BUND forderte bereits 2018 die Behörden auf, mit einem Untersuchungsprogramm, die Eintragspfade von Arzneiwirkstoffen und antibiotikaresistenten Bakterien in die Agger aufzuklären, vor allem den Einfluss der an die Kläranlage angeschlossenen Kliniken. Ziel sollten sinnvolle und wirksame Maßnahmen zur Verringerung der Belastung sein. Obwohl nicht als Badegewässer ausgewiesen, wird auch die Agger zur Naherholung, zum Angeln und auch zur gelegentlichen Erfrischung an heißen Sommertagen genutzt – und gerade dann ist die Gefahr von Infektionen oder der Übertragung solcher Keime auf andere Menschen hoch. Die vom Umweltministerium angekündigten NRW-weiten Untersuchungen hält der BUND für wichtig, aber nicht ausreichend. Diese geben nur einen groben Überblick über die landesweite Problematik und können im konkreten Fall nur bedingt weiterhelfen.
In der Zwischenzeit fand ein Gespräch des BUND mit der Leitung des für die Unterhaltung der Agger und die Kläranlage Engelskirchen zuständigen Aggerverbandes statt. Hierbei wurden die BUND-Ergebnisse durch frühere, vom Verband veranlasste, Untersuchungen bestätigt. Allerdings hatte der Verband nicht nach Antibiotika gesucht.
Auch die Problematik der Resistenzbildung im Gewässern nimmt der Verband ernst, will aber kein „kostspieliges“ Sondermessprogramm auflegen, sondern aktiv das aktuell anlaufende Messprogramm des Landes unterstützen. Hierbei werden nach Aussagen des Landes die BUND Messstellen einbezogen. Aggerverband und BUND haben sich darauf verständigt, auf Basis dieser Ergebnisse dann über Konsequenzen und weitere Maßnahmen zu beraten. (Paul Kröfges)
Artikel aus dem Pharma-Brief 3/2019, S.6
Bild © BUND NRW
[2] Dabei werden nach Vorgaben der Kommission für Krankenhaushygiene und Infektionsprävention beim Robert Koch Institut bis zu 19 Antibiotika, variiert je nach festgestellter Bakterienart, getestet
[3] Alle Ergebnisse: www.bund-nrw.de/presse/detail/news/bund-weist-multiresistente-keime-in-gewaessern-nach
[4] Abwasserkanäle, in denen Schmutzwasser und Regenwasser gemeinsam ins Klärwerk fließen
Antibiotika-Resistenzen
Neues Projekt beleuchtet länderspezifische Facetten
Resistente Erreger verbreiten sich rund um den Globus. Bei Armutskrankheiten wie Tuberkulose und nicht zuletzt im Bereich der Mutter-Kind-Gesundheit sorgen sie für massive Behandlungsprobleme, eine Explosion der Gesundheitskosten und hohe Sterberaten. Die BUKO Pharma-Kampagne plant zahlreiche Aktivitäten, um dieser Thematik mehr öffentliche Aufmerksamkeit zu verschaffen.
Das drohende Zukunftsszenario eines post-antibiotischen Zeitalters bezeichnete die WHO schon 2015 als „globale Gesundheitskrise“. Die Folgen sind verheerend, vor allem in armen Ländern. Einerseits leiden Menschen, denen es an guten Lebensbedingungen, sauberem Trinkwasser oder sanitärer Versorgung mangelt, wesentlich häufiger unter Infektionen. Andererseits sind Therapien gegen resistente Erreger oft nicht verfügbar oder unbezahlbar. Zudem greifen PatientInnen, die für eine ärztliche Beratung selbst aufkommen oder dafür beschwerliche Wege auf sich nehmen müssen, häufig ohne Diagnose zu Antibiotika. Das gilt besonders, wenn diese Mittel rezeptfrei und billig zu haben sind. Der übermäßige und unsachgemäße Gebrauch von Antibiotika beschleunigt wiederum die Resistenz-Entwicklung erheblich.
Das Beispiel Gonorrhö
Antibiotika-Resistenzen (ABR) führen dazu, dass bisher leicht therapierbare Erkrankungen plötzlich unbeherrschbar werden. Ein Blick auf das Beispiel Gonorrhö verdeutlicht die dramatische Situation: Mit 78 Millionen Erkrankungen jährlich ist sie weltweit die zweithäufigste sexuell übertragbare Erkrankung. Die höchsten Infektionsraten verzeichnet der afrikanische Kontinent. Die von der Krankheit hervorgerufenen Entzündungen können besonders für Frauen gefährliche Konsequenzen haben, zudem erhöhen sie das Risiko einer HIV-Infektion. Resistente Erregerstämme machen die Behandlung von Gonorrhö vielerorts inzwischen nahezu unmöglich.[1] In Großbritannien sorgte im März 2018 ein Fall für Aufsehen, der erstmalig Resistenzen gegen beide Antibiotika der Standard-Behandlung aufwies. Die Ansteckung war in Südostasien erfolgt.[2]
Steigender Verbrauch ein Problem
Der Verbrauch von Antibiotika steigt weltweit kontinuierlich an. Eine 2018 veröffentlichte Studie stellte fest, dass der Verkauf zwischen 2000 und 2015 um rund 65% zugenommen hat. In Ländern geringen und mittleren Einkommens gab es einen Anstieg um 114%. Indien stach dabei besonders hervor.[3] In Deutschland zeigen Analysen der Krankenkassen, dass Antibiotika auch bei uns häufig falsch und unnötig verordnet werden.[4],[5]
Eine zentrale Rolle bei der Entwicklung resistenter Keime spielt auch die Landwirtschaft. Der massive Einsatz von Antibiotika bei der Tiermast bringt längst auch Reservepräparate an den Rand ihrer Wirksamkeit. 2015 sorgte der Fund eines neuen Resistenz-Gens bei Schweinen und Hühnern in China für Entsetzen. Selbst Colistin, ein wichtiges Mittel der letzten Reserve, wirkte bei Tieren und Menschen nicht mehr. Nur ein Jahr später wurde das leicht zwischen Bakterien übertragbare Gen auch in Deutschland und anderen europäischen Staaten bei Patienten nachgewiesen.[6],[7]
Weil die Nachfrage nach billigem Fleisch kontinuierlich steigt, nimmt die Massentierhaltung in armen Ländern rasant zu. Die hochgezüchteten Rassen sind jedoch selten an die klimatischen Bedingungen in Südländern angepasst. Die Tiere erkranken öfter und werden häufiger mit Antibiotika behandelt. Auch als Masthilfe bzw. Wachstumsbeschleuniger werden Antibiotika eingesetzt. Die dadurch entstehenden Resistenzen werden durch die Import- und Export-Beziehungen im Fleischhandel globalisiert.
Resistente Keime in Flüssen
Nicht zuletzt birgt die Herstellung von Antibiotika, die oft in Indien und anderen Ländern des globalen Südens erfolgt, gravierende Probleme mit sich. Ende 2017 stieß ein Team deutscher Journalisten bei Wasserproben in Hyderabad/Indien, woher auch fast alle großen deutschen Pharmahersteller Antibiotika beziehen, auf extrem hohe Konzentrationen antibiotischer Wirkstoffe in Gewässern sowie im Grund- und Trinkwasser (Pharma-Brief 5-6, 2017, S. 1.) Die so befeuerte Entwicklung von Resistenzen stellt nicht nur ein lokales Gesundheitsrisiko dar. Denn resistente Erreger kennen keine Grenzen und breiten sich z.B. durch Tourismus weltweit aus. Eine Leipziger Studie fand bei über 70 Prozent der Indien-Reisenden nach ihrer Rückkehr resistente Erreger. Bei Rückkehrenden aus Südostasien waren es fast 50 Prozent.[8]
In jüngster Zeit sind Antibiotika-resistente Erreger in deutschen Gewässern und Badeseen in den Fokus gerückt. Im Februar 2018 fand sich bei Wassertests an verschiedenen Bächen und Seen Niedersachsens eine Vielzahl resistenter Erreger. (Der Pharma-Brief berichtete, 2/2018, S. 2) Antibiotische Wirkstoffe aus der Tierhaltung sind dafür maßgeblich verantwortlich. Sie gelangen u.a. über die ausgebrachte Gülle in Boden und Gewässer. Aber auch Abwässer aus Krankenhäusern und Pflegeheimen weisen hohe Konzentrationen antibiotischer Rückstände auf. Die Kläranlagen sind derzeit technisch nicht dafür ausgerüstet, solche Rückstände von Medikamenten aus dem Wasser herauszufiltern.
Was tut die Pharma-Kampagne?
Die BUKO Pharma-Kampagne nimmt die globale Resistenz-Problematik mit einem neuen Projekt unter die Lupe: Unter dem Titel „Antibiotika-Resistenzen in Nord und Süd - Globale Herausforderungen erkennen, lokale Handlungsoptionen fördern“ werden wir gemeinsam mit Partnerorganisationen in Indien, Tansania und Südafrika eine Datenerhebung durchführen. Im Frühsommer 2020 wollen wir die Ergebnisse und Analysen in einer Broschüre veröffentlichen und dabei auch auf die Situation in Deutschland Bezug nehmen.
Außerdem soll eine Wanderausstellung die komplexe Problematik ansprechend und authentisch vermitteln. Großformatige Bildtafeln, interessante Texte und multimediale Elemente sollen die Resistenzprobleme in verschiedensten Teilen der Welt abbilden, Ursachen aufzeigen und lokale Lösungsstrategien und Handlungsansätze vorstellen. Die Ausstellung wird sich an kritische VerbraucherInnen sowie Beschäftigte in der Landwirtschaft und im Gesundheitswesen richten und soll ab Sommer 2020 im Rahmen von Messen, Kongressen und Veranstaltungen gezeigt werden.
Zusätzlich ist im kommenden Jahr eine zweiwöchige bundesweite Theatertournee geplant, die die Inhalte der Ausstellung aufgreift. So wollen wir der Thematik eine möglichst breite Öffentlichkeit verschaffen, zur politischen Meinungsbildung beitragen und zugleich kommende Ausstellungstermine effektvoll bewerben. Nicht zuletzt sollen ein großes Symposium (2021), gezielte Advocacy- und Öffentlichkeitsarbeit sowie interdisziplinäre Fachgespräche die Probleme bekannter machen und zu einer Vermeidung von Antibiotika-Resistenzen sowie zu entsprechenden gesellschaftlichen Veränderungen in Deutschland beitragen. Das von der Nordrhein-Westfälischen Stiftung Umwelt und Entwicklung geförderte Projekt startete im Juni 2019, Aktivitäten sind bis Ende 2021 geplant.
Artikel aus dem Pharma-Brief 3/2019, S.5
[1] WHO (2017) Antibiotic-resistant gonorrhoea on the rise, new drugs needed. Meldung vom 7.Juli www.who.int/news-room/detail/07-07-2017-antibiotic-resistant-gonorrhoea-on-the-rise-new-drugs-needed [Zugriff 24.6.19]
[2] Gallagher J (2018) ‚World‘s worst‘ super-gonorrhoea man cured. BBC news v. 20.April www.bbc.com/news/health-43840505 [Zugriff 24.6.19]
[3] Klein EY et al. (2018) Global increase and geographic convergence in antibiotic consumption between 2000 and 2015.PNAS; 115, p E3463 https://doi.org/10.1073/pnas.1717295115
[4] Ärzteblatt (2016) BKK-Studie: Ärzte verschreiben Antibiotika oft auf Verdacht. 5.August www.aerzteblatt.de/nachrichten/69907/BKK-Studie-Aerzte-verschreiben-Antibiotika-oft-auf-Verdacht [Zugriff 24.6.19]
[5] TK (2018) Berliner Ärzte verschreiben am seltensten Antibiotika bei Erkältungen. Pressemitteilung vom 15. Oktober www.tk.de/presse/themen/arzneimittel/antibiotika-2044926 [Zugriff 24.6.19]
[6] RKI (2016) Colistin-Resistenz bei Gram-negativen Bakterien – die Situation in Deutschland. Epidemiologisches Bulletin. S. 513
[7] Reardon S (2017) Resistance to last-ditch antibiotic has spread farther than anticipated. Nature, 12.6. www.nature.com/news/resistance-to-last-ditch-antibiotic-has-spread-farther-than-anticipated-1.22140 [Zugriff 24.6.19]
[8] Lübbert C et al. (2015) Coloni Colonization with extended-spectrum beta-lactamase-producing and carbapenemase-producing Enterobacteriaceae in international travelers returning to Germany. International Journal of Medical Microbiology; 305, p 148 https://doi.org/10.1016/j.ijmm.2014.12.001