
2018-artikel
Zwischen Kommerz und Transparenz
Update: europäische Nutzenbewertung
Der EU-Vorschlag zur Nutzenbewertung für Arzneimittel befindet sich mitten in der parlamentarischen Debatte und auch der Ministerrat beginnt sich eine Meinung zu bilden. Wie ist der Stand?
Anfang des Jahres legte die EU-Kommission einen kontroversen Verordnungsentwurf für eine einheitliche europäische Nutzenbewertung (HTA) vor (wir berichteten[1]). Wichtigster Kritikpunkt war das im Artikel 8 enthaltene Verbot nationaler Bewertungen neuer Arzneimittel, verbunden mit einer verpflichtenden Übernahme der EU-Bewertung.
Gegen diese Regelung hat sich am deutlichsten der Ministerrat positioniert. Am 9. Juli hatte die EU-Kommission zu einer Diskussion über die Zukunft von HTA nach Brüssel eingeladen. Für die österreichische Ratspräsidentschaft machte Clemens Auer dort sehr deutlich, dass die Gesetzgebung am Artikel 8 scheitern würde, wenn dort nicht die verpflichtende Übernahme der EU-Bewertungen gestrichen würde. Eine Mehrheit der Mitgliedsstaaten sei gegen diesen Zwang. Außerdem mahnte er ein transparentes Verfahren an.
Im EU-Parlament sind zwei Ausschüsse mit dem Verordnungsentwurf befasst. Der Ausschuss für den internen Markt und Verbraucherschutz hat sich bereits auf Kompromissformulierungen verständigt.[2]
Positiv zu vermerken sind mehrere Formulierungen, die mehr Transparenz im Verfahren einfordern und eine Veröffentlichung der Studienergebnisse, auf denen die Bewertung beruht. Eine sogenannte Koordinierungsgruppe aus Fachleuten der Mitgliedsstaaten, spielt in der Organisation des geplanten Bewertungsverfahrens eine zentrale Rolle. Allerdings liegt sie im Verordnungsentwurf am Gängelband der Kommission, die vieles im Ablauf des Verfahrens selbst festlegen möchte. Das sehen die ParlamentarierInnen anders. Sie wollen, dass die Koordinierungsgruppe die Methoden der Bewertung festlegt, und dass dies in einem transparenten Prozess geschehen soll.
Negativ fällt auf, dass der Hersteller während des Verfahrens das Recht erhalten soll, Einwendungen gegen die EU-Bewertung zu erheben. Diese dürfen von der Koordinierungsgruppe nur mit ausführlicher Begründung abgelehnt werden. Auch die Aufweichung der Anforderungen an die Evidenz bei neuen Therapieformen und Medikamenten gegen seltene Krankheiten sind kontraproduktiv. Sie fördern die Einführung von zu wenig erprobten Methoden.
ENVI-Ausschuss
Der Ausschuss für Umweltfragen, öffentliche Gesundheit und Lebensmittelsicherheit (ENVI) hat sich noch nicht auf Kompromisse für die zahllosen Änderungsanträge geeinigt. Es kursiert aber der Vorschlag, die Auswertung von der Bewertung der Evidenz abzutrennen. Die Auswertung der vorhandenen Studien soll EU-weit geschehen, die Bewertung der wissenschaftlichen Fakten soll nationale Zuständigkeit bleiben.
Deutsch-französischer Vorstoß
Damit wäre der ENVI-Ausschuss in dieser Frage schon nah an einem informellen Vorschlag von Deutschland und Frankreich, der uns vorliegt. Damit wollen die beiden Länder die Debatte um den EU-HTA konstruktiv voranbringen und nennen einige Eckpunkte, die das Verfahren verbessern könnten.
So soll sich die EU auf eine rein deskriptive wissenschaftliche Analyse der vorgelegten Studiendaten beschränken. Wenn über ein Verfahren keine Einigkeit erzielt wird, können Mitgliedstaaten im Einzelfall aus der gemeinsamen Bewertung aussteigen. Die Bewertung des Ausmaßes eines Zusatznutzens soll Privileg der Mitgliedsstaaten bleiben. Zusätzliche wissenschaftliche Auswertungen durch Mitgliedsstaaten müssten möglich bleiben, falls das wegen des nationalen Versorgungskontextes sinnvoll ist.
Die Kriterien für die wissenschaftliche Auswertung der Studien sollten nicht nachträglich von der Kommission selbst bestimmt werden können, sondern müssten bereits im Gesetzestext festgelegt werden. Dahinter steht die Befürchtung, dass statt patientenrelevanter Ergebnisse wie Sterblichkeit oder Linderung der Symptome, möglicherweise Surrogatparameter wie Blutzuckerwerte oder Tumorwachstum als bedeutsamer Vorteil dargestellt werden könnten.
Das Verfahren der wissenschaftlichen Auswertung müsse ganz in der Hand der Koordinierungsgruppe bleiben. Die Kommission soll sich auf die reine Rechtsaufsicht beschränken und – anders als im ursprünglichen Verordnungsentwurf – keine inhaltlichen Einflussmöglichkeiten erhalten.
Ungesunde Hast
Ein Dogma, das Industrie und Kommission gezielt gefördert haben, wurde bislang nicht ernstlich in Frage gestellt: Die angeblich so wichtigen neuen Therapien müssten den PatientInnen noch schneller zur Verfügung stehen. Die Kommission sieht im Verordnungsentwurf nur einen Zeitraum von 67 Tagen für die Bewertung vor, also die Zeitspanne zwischen einer Zulassungsempfehlung der EMA und der Marktzulassung durch die Kommission. Dieser Zeitraum ist für eine seriöse wissenschaftliche Auswertung der Evidenz zu kurz. Bislang begannen Nutzenbewertungen erst nach der Marktzulassung. Der frühere Beginn bedeutet zudem, dass noch weniger Daten aus klinischen Studien vorliegen.
Falsches Instrument
Ein generelles Missverständnis, dem die ParlamentarierInnen aufgesessen sind, ist die Annahme, dass eine einheitliche EU-Bewertung des Zusatznutzens den Zugang zu neuen Arzneimitteln und Medizinprodukten in den Mitgliedsstaaten verbessern werde. Die hohen Kosten für viele neue Medikamente werden das auch künftig verhindern. Außerdem ist es schon jetzt so, dass die Hersteller etliche ihrer hochpreisigen Neuheiten in den ärmeren Mitgliedsstaaten gar nicht vermarkten. Daran kann ein EU-HTA nichts ändern. Allerdings hätte das Parlament die Macht, ein Gesetz auf den Weg zu bringen, dass die Marktzulassung mit der Verpflichtung verbindet, ein Medikament EU-weit anzubieten. Und auch bei der Preisregulierung sind schärfere Regeln durchaus vorstellbar.
Die Debatte über den EU-HTA Ende August wird nach der Sommerpause des EU-Parlaments weitergehen. (JS)
Artikel aus dem Pharma-Brief 6/2018, S. 5
[1] Pharma-Brief (2018) Wunschkonzert für Hersteller. Nr. 3, S. 1
[2] Committee on the Internal Market and Consumer Protection (2018) 2018/0018(COD) Compromise amendments 1 – 21 www.europarl.europa.eu/meetdocs/2014_2019/plmrep/COMMITTEES/IMCO/DV/2018/07-11/CA_health_EN.pdf [Zugriff 19.7.2018]
Zündende Ideen für eine global gerechte Versorgung
Jahresrückblick 2017
Drei große Jahresprojekte mit zahlreichen Aktivitäten prägten die Arbeit der BUKO Pharma-Kampagne im vergangenen Jahr. Gemeinsam mit unseren Netzwerk-Partnern und internationalen ExpertInnen entwickelten wir Strategien für eine bessere globale Gesundheit und leisteten zu unseren Themen eine hartnäckige Advocacy- und professionelle Presse-Arbeit.
Unser besonderes Augenmerk lag 2017 auf der weltweiten Zunahme von Antibiotika-Resistenzen. Wir machten uns mit unserer politischen Arbeit für tragfähige Handlungsalternativen stark und für strukturelle Veränderungen, die dazu beitragen können, Resistenzen zu vermeiden und die Forschung zu antibiotischen Wirkstoffen anzukurbeln. Im Januar 2017 luden wir GesundheitswissenschaftlerInnen und Fachleute aus Human- und Veterinärmedizin zu einer Konferenz nach Bielefeld ein, um gemeinsam wirksame Strategien zu entwickeln. Unsere Broschüre „Wettlauf gegen die Zeit“ fasst die Diskussionsergebnisse der Tagung ansprechend und informativ zusammen.
Advocacy zum G-20 Gipfel
Insbesondere im Vorfeld des G-20-Gipfels führten wir zahlreiche Gespräche mit politischen EntscheidungsträgerInnen und vielen anderen Akteuren – so etwa beim Runden Tisch Gesundheit von BMZ / BMBF im Januar in Bonn. Wir zeigten gefährliche Forschungs-Engpässe bei antibiotischen Wirkstoffen auf und trugen dazu bei, das Thema in Hamburg und anderswo auf die politische Tagesordnung zu bringen.
Finanzspritze für die Antibiotika-Forschung
Erfreulicherweise sagte Bundesgesundheitsminister Hermann Gröhe im September über 50 Millionen Euro für das Forschungsprojekt GARDP zu, einer von der WHO und der Drugs for Neglected Diseases (DNDi) initiierten Partnerschaft zur Antibiotika-Forschung.
Unser kritisch-konstruktiver Blick auf die deutsche Politik fand nicht zuletzt Eingang in einen Schattenbericht über Deutschlands Beitrag zu den Nachhaltigkeitszielen: Im Bericht „Großbaustelle Nachhaltigkeit - Deutschland und die globale Nachhaltigkeitsagenda 2017“ war unser Mitarbeiter Christian Wagner-Ahlfs Mitautor eines Kapitels zu Antibiotika-Resistenzen als globaler Herausforderung.
Bildungsarbeit zu Tuberkulose
Weiterhin stand die Armutskrankheit Tuberkulose im Fokus unserer Projektarbeit: Die neue Unterrichtsmappe „Da kriegste die Motten!“ wurde intensiv beworben und ist inzwischen in gut einem Dutzend Online-Portalen zum Globalen Lernen und Suchmaschinen für Lehrmaterialien aufzufinden. Die Mappe wurde an knapp 70 Mediotheken und Online-Portale sowie an rund 100 Lehrkräfte und MultiplikatorInnen entwicklungspolitischer Bildungsarbeit verschickt. Sie steht auf unserer Website zum kostenlosen Download bereit und wird dort durch Online-Angebote, Filmtipps, weiterführende Literatur und Exkursionsvorschläge sinnvoll ergänzt. Bei zahlreichen Fortbildungen und Fachtreffen haben wir unsere Materialien vorgestellt und bei fünf Unterrichtsbesuchen an weiterführenden Schulen und Berufsschulen eingesetzt.
Straßentheater
Nicht zuletzt führte unsere Theatergruppe Schluck&weg die Probleme bei der Tuberkulose-Bekämpfung eindrucksvoll vor Augen. Die Tournee erreichte mit 15 Auftritten an Schulen über 1.100 SchülerInnen. Zusätzlich sahen weit über 2.000 PassantInnen. die Vorführungen auf der Straße. Eine filmische Dokumentation des Theatertücks „Schiller und die Gesundheitsräuber“ ist online verfügbar und wurde auch auf der Jugendwebsite EineWeltblabla eingestellt.
Gerechte Lizenzen
Mit einem 2017 gestarteten Projekt zur sozial gerechten Patentverwertung tragen wir dazu bei, innovative Forschungs-Produkte im globalen Süden besser verfügbar machen. Gemeinsam mit einer Juristin entwickelten wir Vertragsbausteine, um alternativen Lizenzverträgen im Bereich der öffentlichen Forschung den Weg zu ebnen. Bei sechs öffentlichen Veranstaltungen an Universitäten und sechs Fachseminaren mit Lizenz-Verwertungsagenturen stellten wir gerechte Lizenzmodelle vor.
Nicht zuletzt haben wir zahlreiche WissenschaftlerInnen zu einer sozial gerechten Verwertung ihrer Forschungsergebnisse beraten.
Veranstaltungen und Vorträge
Die Pharma-Kampagne war bei rund 70 Veranstaltungen, Fachtreffen und Konferenzen im In- und Ausland vertreten, häufig mit Vorträgen, dezidierten Stellungnahmen oder auf dem Podium. Allein zehn Vorlesungen und Fachvorträge hielten wir an deutschen Universitäten und auch bei vielen Veranstaltungen unserer Bündnispartner war unsere Expertise gefragt: So referierten wir beim 15jährigen Jubiläum des Aktionsbündnis gegen Aids, bei einer Veranstaltung von Brot für die Welt mit dem Titel „How will we achieve affordable medicines for all?“, bei einer Tagung der Plattform Globale Gesundheit oder auch bei einem Europatreffen der EU for Health Alliance. Weiterhin unterstützen wir das IPPNW-Studierendentreffen mit einem Workshop zur Rolle der Pharmaindustrie in der globalen Arzneimittelversorgung.
Pressearbeit
Im vergangenen Jahr publizierten wir zehn Pharma-Briefe und erreichten mit unserer Presse- und Öffentlichkeitsarbeit ein beachtliches Medienecho. Über unsere Themen und Aktivitäten berichteten u.a. Arte, Frontal 21, Report Mainz, Monitor, verschiedene ARD-Radiosendungen sowie der Deutschlandfunk, aber auch die Zeit, der Evangelische Pressedienst, die Wirtschaftswoche sowie die Zeitschriften Gesundheitswesen und Dr. med Mabuse. Wir standen bei 62 Anfragen von JournalistInnen Rede und Antwort – lieferten stichhaltige Informationen, führten Interviews und Hintergrundgespräche oder vermittelten Kontakte zu ExpertInnen im In- und Ausland. Und auch über soziale Medien haben wir unsere Themen intensiv kommuniziert: Wir verfassten 191 Einträge bei Facebook und verschickten 251 Tweets zu aktuellen politischen Entwicklungen. Dabei standen vor allem unsere Schwerpunktthemen Tuberkulose und Antibiotika-Resistenzen, aber auch die Beeinflussung der WHO durch die Gates-Stiftung im Fokus.
Neue Website geht an den Start
Auf unserer Website stellten wir 61 aktuelle Meldungen bereit und wollen unser Online-Informationsangebot künftig noch weiter ausbauen. Im Herbst haben wir deshalb mit einem Relaunch unserer Website begonnen: Unser Internetauftritt soll nutzerInnen-freundlicher werden, unsere Arbeitsschwerpunkte übersichtlicher präsentieren und auch kompatibel für Smartphones sein. Die neue Homepage geht demnächst an den Start.
Wir bedanken uns bei allen, die unsere Arbeit durch ihre Spende, ihre fachliche Unterstützung oder auch durch ihr freiwilliges Engagement unterstützt haben. (CJ)
Artikel aus dem Pharma-Brief 2/2018, S. 6
Bild © Tim Rohlfs
Zugang zu Medikamenten in Europa verbessern
Forschungspolitische Wende nötig
Die Europäische Union muss die Menschen in den Mittelpunkt ihrer Forschungspolitik stellen. Das fordern 25 Organisationen, darunter auch die BUKO Pharma-Kampagne, in einem Manifest. Viele Gesundheitssysteme in der EU leiden unter den Folgen einer einseitigen Industriepolitik im pharmazeutischen Sektor. Diese zielt fast ausschließlich auf Wirtschaftswachstum und Gewinnmaximierung, statt auf die optimale Versorgung der Bevölkerung mit guten und bezahlbaren Medikamenten. Bessere Regeln für Forschung und Entwicklung können eine Kehrtwende einleiten.
Das Manifest fordert einen „Public return on public investment“. Steuergelder, die in biomedizinische Forschung und Entwicklung investiert werden, müssen den größtmöglichen Nutzen für die Gesellschaft bringen. Hier haben die bisherigen forschungspolitischen Programme nach wie vor große Lücken.
Deutsche Forschungspolitik
So enthält das im November beschlossene „BMBF Rahmenprogramm zur Gesundheitsforschung“[1] des Bundesministeriums für Bildung und Forschung zwar viele wichtige Elemente: internationale Vernetzung der Forschung, non-profit-Kooperationen zu vernachlässigten Krankheiten, staatliches Engagement für die Entwicklung neuer Antibiotika. Doch generell wird das Ziel der „Translation“, also Forschungsergebnisse in die Anwendung zu bringen, im Sinne einer Industrieförderung ausgelegt. Mit keinem Wort wird erwähnt, dass neue Therapien oft mit unsinnig hohen Kosten verkauft werden. Mit keinem Wort wird erwähnt, dass öffentliche finanzielle Förderung an bestimmte Bedingungen gekoppelt werden könnte.
Europawahl 2019: Chance für Richtungswechsel
Auch die europäische Forschungsförderung ist hier noch viel zu zögerlich. Derzeit wird das nächste Forschungsrahmenprogramm „Horizon Europe“ verhandelt, das ab 2021 die Regeln für die Gesundheitsforschung festlegen wird. Es ist zu befürchten, dass trotz vielfacher Forderungen keine Regeln für den Zugang zu Produkten aus öffentlicher Forschung aufgenommen werden.
Da im Mai 2019 das Europäische Parlament neu gewählt wird, besteht Hoffnung auf Nachbesserung. Das nachfolgend abgedruckte Manifest ist somit ein Appell an zukünftige ParlamentarierInnen, sich stärker darum zu kümmern, dass öffentliche Forschung in bezahlbare Produkte mündet. (CW)
Artikel aus dem Pharma-Brief 10/2018, S. 6
[1] BMBF (2018) Rahmenprogramm Gesundheitsforschung der Bundesregierung www.gesundheitsforschung-bmbf.de/files/Rahmenprogramm_Gesundheitsforschung_nicht%20_barrierefrei.pdf
Zu viel Geld für Arzneimittel
Arzneiverordnungsreport 2018 (AVR)
Jedes Jahr bietet der AVR eine umfassende Analyse der ärztlichen Verordnungen zu Lasten der Krankenkassen. Die Kostensteigerungen setzen sich fort, es gibt aber auch kleine Lichtblicke.
Für Medikamente mussten die gesetzlichen Krankenkassen 2017 knapp 40 Milliarden aus den Versichertenbeiträgen aufwenden. Dazu kommen noch die Ausgaben für Medikamente in Krankenhäusern, die im AVR aber nicht dargestellt werden können. Damit sind die Arzneimittelausgaben gegenüber dem Vorjahr um 3,7% gestiegen.[1]
Die Steigerung der Arzneimittelkosten liegt deutlich über den Verbraucherpreisen, die im gleichen Zeitraum nur um 1,7% stiegen. Der Trend bleibt also bedenklich. Vor allem, da 2017 sogar etwas weniger Rezepte ausgestellt wurden als im Vorjahr. Die höheren Kosten erklären sich erneut mit den rasch wachsenden Preisen von patentgeschützten Arzneimitteln. Während patentgeschützte Mittel im Schnitt 2.500 € pro Jahr kosteten, lagen die Preise von Neueinführungen 2017 deutlich höher. Von den 34 neuen Wirkstoffen hatten 24 Jahrestherapiekosten von über 20.000 € pro PatientIn, 9 der 10 Krebsmedikamente kosteten sogar über 60.000 €.
Die Kostensteigerungen wären noch größer, wenn nicht kontinuierlich weniger der teuren Produkte verschrieben würden. Gab es 2008 noch 68 Millionen Rezepte für patentgeschützte Arzneimittel, fiel die Zahl bis 2017 auf 39 Millionen Rezepte. Im gleichen Zeitraum stiegen die Ausgaben für diese Mittel aber von 11,1 Mrd. € auf 18,5 Mrd. €.
Nutzenbewertung dämpft Preise wenig
Auch das AMNOG,[2] das seit 2011 zwingend eine Nutzenbewertung für neue Medikamente mit anschließenden Preisverhandlungen vorschreibt, hat den Trend nicht umkehren können. Zwar wurden gewisse Einsparungen gegenüber den ursprünglichen Einführungspreisen erzielt, aber der Steuerungseffekt blieb mäßig. Das zeigt ein Kapitel zum Verordnungsverhalten bei AMNOG-Arzneimitteln. So werden viele Rezepte für Medikamente ausgestellt, die keinen Zusatznutzen haben oder nur in Teilindikationen (etwas) besser waren. Diese beiden Gruppen machen zusammen mit Abstand den größten Teil der Kosten aus.
Ältere patentgeschützte Arzneimittel tragen ebenfalls zu den hohen Kosten bei – oft fehlt aber der (Zusatz-)nutzen. Sie dürfen aber im AMNOG-Verfahren nach einer Gesetzesänderung seit 2014 nicht mehr auf ihren Nutzen überprüft werden – ein Erfolg der Pharmalobby.
Sonderkapitel
Immer wieder gibt es im AVR besondere Analysen, so auch in diesem Jahr. Ein Kapitel zu den unterschiedlichen europäischen Zulassungsverfahren bringt mehr Klarheit in die zahlreichen Pfade zum Marktzugang. Dabei werden die Unwägbarkeiten der beschleunigten Zulassungswege gut deutlich: Die Ungewissheit, ob ein solcher Schnellschuss den PatientInnen überhaupt hilft, kann beträchtlich sein. Und sie macht die Bewertung des Nutzens extrem schwierig, weil zuverlässige Daten aus kontrollierten klinischen Studien oft fehlen.
Ein besonders problematischer Bereich sind die sogenannten Orphan-Drugs, Arzneimittel für seltene Erkrankungen. Für sie gelten niedrigere Zulassungsanforderungen und eine längere Marktexklusivität. Das entdecken immer mehr Firmen als lukratives Geschäftsfeld. Durch die Identifizierung von Biomarkern – vor allem bei Krebserkrankungen – lassen sich immer kleinere PatientInnengruppen definieren. So nimmt – nicht zuletzt wegen der relativ großzügigen Obergrenze der PatientInnenzahl bei der Definition von Orphan-Drugs, die Zahl der Waisenmedikamente bei den Zulassungen schnell zu. Diese Mittel sind in der Regel extrem teuer und werden oft außerhalb der eigentlich zugelassenen engen Indikation eingesetzt.
Biosimilars
Dem Thema Biosimilars als generische Alternative zu Biologika ist ein eigenes Kapitel gewidmet. Aufgrund des biotechnologischen Herstellungsprozesses in lebenden, gentechnisch veränderten Organismen, der aufgrund von Eigentumsrechten für jeden Hersteller verschieden ist, sind Biosimilars nur sehr ähnlich, aber im Gegensatz zu chemisch hergestellten Arzneimitteln, niemals völlig identisch. Sie müssen jedoch in wesentlichen Strukturmerkmalen gleich sein und gegen das Biologikum in einer klinischen Studie getestet werden. Nur wenn sich keine Unterschiede in der Wirksamkeit und Sicherheit zeigen, werden sie durch die europäische Behörde EMA zugelassen.
Trotzdem werden sie viel zu wenig verwendet. Allerdings tragen auch die Hersteller von Biosimilars zu den hohen Kosten bei, weil sie zunehmend ihre Produkte kaum billiger anbieten als das Original-Biologikum.
Breiter Überblick
Mit zwei Dritteln machen – wie gewohnt – die Analysen der Verschreibungen in verschiedenen Indikationsgruppen den größten Teil des Buches aus. Sie bleiben nicht bei den Zahlen stehen, sondern bieten auch eine Bewertung unter klinisch-pharmakologischen Gesichtspunkten. Es wird bei vielen Erkrankungen deutlich, dass bei der Rationalität des Verschreibungsverhaltens noch deutlich Luft nach oben ist.
Gefühlt fast so schwer wie ein Ziegelstein ist der neue AVR – aber die gut 900 Seiten sind auch vollgepackt mit spannenden Daten, und für alle, die sich intensiver mit dem deutschen Arzneimittelmarkt auseinandersetzen wollen, eine unentbehrliche Quelle. (JS)
Artikel aus dem Pharma-Brief 10/2018, S. 4
Bild © Schwabe U et al. (Hrsg.) (2018) Arzneiverordnungs-Report 2018. Berlin: Springer. 906 S., 59,99 €, eBook 22,99 €, S. 4
[1] Allerdings hat auch die Zahl der Versicherten um 1,8% auf 71,4 Millionen zugenommen, so dass der reale Pro Kopf-Zuwachs geringfügig niedriger ausfällt.
[2] Arzneimittelmarktneuordnungsgesetz. Pharma-Brief (2012) Wem nützt´s? Nr. 5, S. 7
Wunschkonzert für Hersteller
EU will nationale Nutzenbewertung von Arzneimitteln verbieten
Neue Medikamente sind oft nicht besser als existierende Therapien. Deshalb wird in vielen Ländern seit Jahren der relative Nutzen bewertet und anschließend der Preis für die Neueinführungen ausgehandelt. Seit 2011 gibt es ein entsprechendes Verfahren auch in Deutschland. Die EU-Kommission will solche nationale Bewertungen verbieten und durch ein intransparentes zentrales Verfahren mit niedrigen Standards ersetzen.
Auch wenn Deutschland mit der Nutzenbewertung ein Nachzügler war, das hiesige Verfahren gilt als vorbildlich und transparent (siehe im grauen Kasten). Im Gegensatz zu anderen europäischen Ländern dürfen in Deutschland neue Medikamente sofort ab Zulassung zu Lasten der Kassen verschrieben werden. Andernorts ist das erst nach Ende des Bewertungsverfahrens der Fall – und dort werden längst nicht alle Neueinführungen in die Erstattung aufgenommen. Der Pharmaindustrie sind alle diese Verfahren – entgegen anderslautender Lippenbekenntnisse[1] – ein Dorn im Auge, weil sie die Vermarktungsmöglichkeiten für ihre Produkte einschränken. Seit Jahren drängt sie deshalb auf ein einheitliches EU-weites Verfahren.
Dass die EU-Kommission bereit war, diesem Drängen nachzugeben, zeigte sich schon Ende 2016. Bereits damals warnten wir im Pharma-Brief vor einer Gleichschaltung der Nutzenbewertung.[2] Am 31. Januar 2018 legte die EU-Kommission einen Verordnungsentwurf [3] für eine europäische Arzneimittelbewertung vor.[4] In welche Richtung er weist, lässt schon die Begründung aus der Feder der Generaldirektion für Gesundheit erahnen. Von den Problemen, die das Gesetz lösen soll, werden an erster Stelle: „Hindernisse und Verzerrungen beim Marktzugang“ für Medikamente genannt. Im Vordergrund stehen also die Interessen der Hersteller – und nicht die der Kranken, die gute Arzneimittel brauchen. Doch was hat es mit diesen angeblichen „Verzerrungen“ überhaupt auf sich?
Doppelstandards?
Das vordergründige Argument der Industrie, das die Kommission willig aufgegriffen hat, ist, dass die jeweiligen nationalen HTA-Agenturen[5] beim gleichen Wirkstoff durchaus auch mal zu unterschiedlichen Bewertungen kommen. Doch dafür gibt es nachvollziehbare Gründe.
In England z.B. gibt es eine klare finanzielle Grenze: Der Zugewinn eines gesunden Lebensjahres darf maximal Mehrkosten von 30.000 £ verursachen. Senkt der Hersteller den Preis nicht, darf das Medikament im National Health Service nicht verschrieben werden. Gegenwärtig wird sogar debattiert, die Grenze auf 15.000 £ zu senken.[6] In Deutschland gibt es ein solche Einschränkung überhaupt nicht.
In Schweden werden für die Bewertung regelmäßig nur die (immer unvollständigen) Studienveröffentlichungen in medizinischen Journalen herangezogen. Der Schwedische Rechnungshof kritisiert zudem die zahlreichen Interessenkonflikte der an der Arzneimittelzulassung und -bewertung beteiligten Behörden.[7] In Deutschland werden dagegen umfangreiche Unterlagen eingefordert: „Vorzulegen sind Studienberichte einschließlich Studienprotokollen zu Zulassungsstudien sowie alle im Anwendungsgebiet durchgeführten Studien, die der Zulassungsbehörde übermittelt worden sind.“ [8] Für die MitarbeiterInnen des IQWiG, die die wissenschaftliche Bewertung der Herstellerdossiers durchführen, gelten strenge Regeln für Interessenkonflikte.
Zentralisierung
Kern des Gesetzentwurfs ist ein zentralisiertes europäisches Bewertungsverfahren für neue Arzneimittel. Über deren Nutzen soll ein EU-Gremium mit Vertretern nationaler Behörden entscheiden, und das per einfacher Mehrheit. Das Ergebnis der Bewertung ist dann für alle Mitgliedsstaaten bindend. Allerdings behält sich die EU-Kommission ein Vetorecht vor. Dann muss der Hersteller noch mal angehört werden. Er erhält also eine zweite Chance auf eine bessere Bewertung. Dass die Entscheidung zu industriefreundlich und die Kommission deshalb ein Veto einlegen könnte, ist nicht vorgesehen.
Verpflichtend notwendig?
Ungeachtet massiver Kritik sieht der Gesetzentwurf eine verpflichtende EU-Nutzenbewertung aller neuen Medikamente und die Übernahme dieser Entscheidungen auf nationaler Ebene vor. Die EU-Kommission schreckt dabei selbst vor manipulativen Äußerungen nicht zurück. So kann man in der Pressemitteilung anlässlich der Vorstellung des Entwurfs lesen: „Sehr große Unterstützung findet diese Zusammenarbeit auch bei den Interessenträgern und den Bürgerinnen und Bürgern, die sich an der öffentlichen Konsultation der Kommission beteiligt haben: Fast alle (98 %) erkennen den Nutzen der HTA an, und 87 % befürworten eine Fortsetzung der EU-weiten Zusammenarbeit bei der HTA über 2020 hinaus.“ [9] Das sind die einzigen Zahlen, die genannt werden. Dabei stammten erstens über die Hälfte der Einreichungen bei der Konsultation aus der Pharmaindustrie und zweitens hatte sich trotzdem die Mehrheit gegen das von der Kommission gewählte Modell ausgesprochen.
Auch an internen Warnungen hatte es nicht gefehlt. Der Ausschuss für Regulierungskontrolle der EU-Kommission soll Überregulierung verhindern. Er hat die Ideen der Generaldirektion für Gesundheit gleich zweimal beanstandet und Nachbesserungen verlangt: Es sei nicht überzeugend dargelegt worden, warum eine zentralisierte einheitliche Bewertung der beste Weg sei, die Versorgung mit Arzneimitteln in der EU zu verbessern.[10]
Was ist Nutzen?
Eine wichtige Ursache für unterschiedliche Ergebnisse der nationalen Bewertungen sind die Kriterien für den Nutzen. Reicht es für ein Diabetesmedikament aus, dass es den Blutzucker senkt? Oder muss gezeigt werden, dass die Risiken der Erkrankung sinken – PatientInnen also tatsächlich seltener einen Herzinfarkt bekommen, weniger Amputationen notwendig werden usw. (siehe auch Kasten). Die EU-Kommission hat in ihren Papieren zur Vorbereitung des Gesetzgebungsverfahrens genau so ein Beispiel verwendet, um vermeintlich unverständliche unterschiedliche Bewertungen zwischen Frankreich und Deutschland gegenüber Großbritannien und Schweden zu belegen. Sie spricht nur vage von „unterschiedlichen Anforderungen an die Daten“.[11] Dabei ist der Grund für die unterschiedlichen Bewertungen schlicht, dass sich Frankreich und Deutschland für tatsächlich patientenrelevante Verbesserungen interessieren, in den anderen Ländern aber die Blutzuckersenkung als Kriterium ausreicht.
Außerdem werden hier Äpfel mit Birnen verglichen: In Deutschland kann ein Medikament trotz fehlenden Zusatznutzens weiter verschrieben werden, während eine negative Bewertung in anderen Ländern zum Ausschluss aus der Erstattung führt.
Diese wichtigen Unterschiede adressiert der Gesetzentwurf nicht. Er bleibt absolut vage, welche Kriterien für die Nutzenbewertung gelten sollen. Und es steht zu befürchten, dass am Ende ein fauler Kompromiss stehen wird. Zum Schaden der Kranken, denn die bloße Verbesserung von Laborwerten ist noch lange keine Garantie dafür, dass Menschen gesund werden. Das Diabetesmedikament Rosiglitazon sollte ein warnendes Beispiel sein: Der Wirkstoff senkte effektiv den Blutzucker. Aber statt Erkrankungen zu verhindern, führte er zu 100.000 zusätzlichen Herzinfarkten, bevor er nach zehn Jahren verboten wurde.[12]
Nichts genaues weiß man
Welche Belege die Hersteller im Rahmen der Nutzenbewertung vorweisen müssen, will die Kommission selbst festlegen – aber erst, nachdem das Gesetz verabschiedet ist. EU-Parlament und der Rat sollen also eine Black Box verabschieden. Das ist wirklich ziemlich starker Tobak und wäre ein extremer Rückschritt zu gesetzlich präzise fixierten Verfahren wie in Deutschland (siehe Kasten) und einigen anderen Mitgliedsstaaten.
Vorschnelle Bewertung
Nach den Vorstellungen der Kommission soll die Nutzenbewertung zum Zeitpunkt der Zulassung durch die EMA bereits abgeschlossen sein. Derzeit beginnen die Bewertungen des therapeutischen Mehrwerts regelmäßig mit der Zulassung. Und das ist auch sinnvoll. Denn bis zum Tag der Zulassung steht die genaue Indikation noch nicht fest. Oft weicht die EMA von den Wünschen des Herstellers ab und fasst die Anwendungsbereiche enger. Was genau soll man also vor der Zulassung bewerten? Ein früherer Beginn der Bewertung bedeutet auch, dass noch weniger klinische Studien abgeschlossen sind und die Ergebnisse damit noch unsicherer sind. Das ist keineswegs ein theoretisches Problem, denn die EMA lässt Medikamente oft schon zu, wenn sich in laufenden Studien ein positiver Trend zeigt.[13] Außerdem: Wenn die EMA die Zulassung verweigert, ist die bereits durchgeführte Nutzenbewertung sinnlos.
Den wahren Grund für die geforderten Schnellschüsse in der Nutzenbewertung kann man in den Papieren der Kommission finden. Da wird vorgerechnet, dass jeder Monat, den ein Medikament früher in die Erstattung kommt, 130 Mio. € Mehreinnahmen für den Hersteller bedeutet.[14]
Einfluss der Kommission
In vielen Teilen bleibt der Gesetzentwurf vage. Er wimmelt von Klauseln, die der EU-Kommission das Recht geben, Einzelheiten des Verfahrens nach der Verabschiedung der Verordnung selbst festzulegen. Sie bestimmt, welche „Interessenträger“ im Laufe der Bewertung gehört werden und sie erhält ein Kommentierungsrecht während des laufenden Verfahrens. Die EU-Kommission besitzt außerdem ein Vetorecht gegen die Entscheidungen des Bewertungsgremiums. Dann muss der Hersteller noch einmal angehört werden. Sollte das Bewertungsgremium auch im zweiten Anlauf zu keiner der Kommission genehmen Meinung gelangen, gibt es keine EU-weite Bewertung.
Transparenz: Fehlanzeige
Ein weiterer Schwachpunkt des Gesetzentwurfs ist die fehlende Transparenz des Verfahrens, und es bleibt unklar, wieviel von dem Bewertungsbericht bekannt wird. Sicher ist schon jetzt, dass „alle sensiblen Geschäftsdaten aus dem genehmigten Bericht […] gestrichen werden“ [3] und dass eine Veröffentlichung erst nach Abschluss des Verfahrens stattfindet. Das steht in klarem Gegensatz zur Transparenz des deutschen Verfahrens (siehe Kasten).
Reaktionen
Viele ExpertInnen üben massive Kritik an dem Gesetzentwurf. So schreiben das Deutsche Netzwerk für evidenzbasierte Medizin und HTA.de in einer gemeinsamen Stellungnahme: „[Es werden] durch Wirtschaftsinteressen geprägte Bewertungen durch die EU-Kommission autorisiert. […] Die EU-Kommission behält sich weitreichende Möglichkeiten der Einflussnahme vor. So will sie etwa methodische und prozessbezogene Vorgehensweisen per Rechtsverordnung festlegen und somit Einfluss auf die Bewertungsmethodik nehmen. […] Es fehlt beispielsweise eine Verpflichtung der Industrie zur Bereitstellung einer vollständigen Datengrundlage, die auch unpublizierte Daten umfasst.“ [15]
Der deutsche Bundestag beschloss am 22.3.2018 einstimmig eine Subsidaritätsrüge gegenüber der EU wegen Verletzung des Lissabon-Vertrags. Der Verordnungsentwurf greife „in die rechtlich geschützte Zuständigkeit der Mitgliedstaaten für die Festlegung ihrer Gesundheitspolitik sowie für die Organisation des Gesundheitswesens und medizinischen Versorgung“ ein.[16] Auch Polen, Frankreich und Tschechien erteilten der EU-Kommission eine Rüge. Hätten acht oder mehr Länder protestiert, hätte ein neuer Gesetzentwurf geschrieben werden müssen. Andererseits haben bei der endgültigen Entscheidung über das Gesetz vier Mitgliedsstaaten eine Sperrminorität, die die EU-Verordnung endgültig zu Fall bringen würde.
Sand in die Augen
Mit einem Trick versucht der Gesetzentwurf den Eindruck zu erwecken, dass die im Lissabon-Vertrag der EU garantierte nationale Zuständigkeit für die Gesundheitsversorgung nicht angetastet wird. So soll „nur“ die wissenschaftliche Bewertung zentralisiert werden, die „Bewertung der nichtklinischen (z. B. wirtschaftlichen, sozialen und ethischen) Aspekte einer Gesundheitstechnologie wie auch die Festlegung von Preisen und Erstattungssätzen bleibt weiterhin Sache der einzelnen EU-Mitgliedstaaten.“ [7]
Das ist eine künstliche Trennung. Die Bundesärztekammer merkt in ihrer Stellungnahme dazu an: „Methodisch ist die Trennung in klinische und nichtklinische Dimensionen artifiziell und verkennt, dass die Durchführung eines HTAs bereits zu Beginn ein Gesamtkonzept erfordert, aus dem hervorgehen sollte, auf welche Dimensionen sich die Nutzenbewertung erstrecken wird. Dies bestimmt maßgeblich den Umfang und die Inhalte der durchzuführenden Literatur- bzw. Evidenzrecherchen. […] Auch die Zuordnung, welche Aspekte des Nutzens rein ‚klinisch‘ sind und welche z. B. zur ethischen Kategorie zählen sollen und gesondert zu betrachten wären, ist bei weitem nicht so trennscharf möglich, wie es im Kommissionspapier unterstellt wird.“[17]
… und stattdessen?
Die Bewertung des tatsächlichen Nutzens von Arzneimitteln für PatientInnen ist eine gute Sache. Sie schafft für Kranke und VerschreiberInnen mehr Klarheit, was von den Neuerungen zu erwarten ist und was nicht. Deshalb ist auch eine europäische Zusammenarbeit sinnvoll. Es kommt aber sehr auf das „wie“ an. Es gibt bereits seit Jahren durch EUnetHTA eine freiwillige Zusammenarbeit der Beteiligten aus praktisch allen EU-Mitgliedsstaaten, die auch durchaus schon zu Verbesserungen der nationalen Bewertungsverfahren geführt hat.
Allerdings krankt EUnetHTA an mehreren Problemen: Die Finanzierung stand stets auf wackeligen Beinen und es gab Vorschläge der Kommission, die Arbeit ausgerechnet durch die Beratung der Industrie bei der Entwicklung von neuen Wirkstoffen zu finanzieren. Wie soll da die Unabhängigkeit gewahrt bleiben?
Zwischen den Agenturen in reicheren Mitgliedsstaaten mit hunderten von MitarbeiterInnen und dem Rest von Europa klafft eine nur schwer zu überbrückende Lücke: Denn je höher die wissenschaftlichen Anforderungen an die Bewertung sind, um so arbeitsintensiver wird das Ganze – für kleine Agenturen eine kaum zu bewältigende Aufgabe. Schließlich gibt es bei EUnetHTA ein Problem mit Interessenkonflikten, denn die Anforderungen an die Unabhängigkeit der Mitwirkenden sind keineswegs einheitlich und bei vielen Treffen sitzt die Industrie mit im Boot.
Es käme also erst einmal darauf an, EUnetHTA eine solide Finanzierungsbasis zu bieten und für Unabhängigkeit von der Industrie zu sorgen. Das würde eine konstruktive europäische Zusammenarbeit bei der Bewertung von Arzneimittel fördern. Warum solche Ergebnisse dann allerdings allen Mitgliedsstaaten aufgezwungen werden sollen, das weiß die Kommission allein – Wissenschaft lebt von der Überprüfbarkeit ihrer Ergebnisse.
Realpolitik
Jetzt steht aber erst einmal die Debatte über den Verordnungsentwurf im EU-Parlament an und dann eine Entscheidung des Europäischen Rats. Wir werden die weitere Entwicklung aufmerksam beobachten. (JS)
Artikel aus dem Pharma-Brief 3/2018, S. 1
Bild: Berlaymont, Sitz der EU-Kommission in Brüssel © Jörg Schaaber
[1] “Continued collaboration on how we introduce these new technologies into our healthcare systems will be key. HTA is an important part of that process, informing health care decision makers about the relative value of health technologies.” EFPIA (2018) EFPIA views Commission’s proposals on Health Technology Assessment (HTA) as positive step. 31 Jan.
[2] Pharma-Brief (2016) Gleichschaltung geplant. Nr. 8, S. 5
[3] Eine Verordnung hat unmittelbar EU-weit Gesetzeskraft. Dagegen muss eine EU-Richtlinie in nationale Gesetze umgesetzt werden, wodurch ein gewisser Spielraum entsteht. Im weiteren Text wird EU-Verordnung und Gesetz synonym benutzt.
[4] European Commission (2018) Proposal for a regulation of the European Parliament and of the Council on health technology assessment and amending Directive 2011/24/EU. COM(2018) 51 final http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A52018PC0051
[5] Health Technology Asssessment (HTA), die Bewertung von Gesundheitstechnologien
[6] O’Dowd A (2018) New drugs: patient hope or harm? BMJ; 360, p k1373
[7] Swedish National Audit Office (2016) Safe and effective medicines – how does central government deal with the influence of the pharmaceutical industry? RiR 2016:9
[8] G-BA (2013) Format und Gliederung des Dossiers, einzureichende Unterlagen, Vorgaben für technische Standards. www.g-ba.de/downloads/17-98-3518/2013-04-18_Anl2_1_Erstellung-Einreichung-Dossier.pdf [Zugriff 29.4.2018]
[9] Europäische Kommission (2018) Bewertung von Gesundheitstechnologien in der EU: Kommission schlägt verstärkte Zusammenarbeit der Mitgliedstaaten vor Pressemitteilung 31. Jan. http://europa.eu/rapid/press-release_IP-18-486_de.htm [Zugriff 29.4.2018]
[10] https://ec.europa.eu/health/sites/health/files/technology_assessment/docs/2018_ia_rsbopinion_en.pdf [Zugriff 29.4.2018]
[11] European Commission (2016) Inception Impact Assessment HTA http://ec.europa.eu/smart-regulation/roadmaps/docs/2016_sante_144_health_technology_assessments_en.pdf [Zugriff 29.4.2018]
[12] Pharma-Brief (2010) Absturz überfällig. Rosiglitazon verboten, Nr. 8, S. 7
[13] Pharma-Brief (2017) Viel Lärm um nichts? Streit um Brustkrebsmedikament. Nr. 4, S. 4
[14] European Commission (2018) Impact Assessment. Strengthening of the EU Cooperation on Health Technology Assessment (HTA). SWD(2018) 41 final https://ec.europa.eu/health/sites/health/files/technology_assessment/docs/2018_ia_final_en.pdf
[15] DNEbM und HTA.de (2018) Harmonisierung um jeden Preis? Evidenzbasierte Gesundheitsversorgung geht vor Binnenmarkt und Profitinteresse. Gemeinsame Stellungnahme 14. April. www.ebm-netzwerk.de/aktuelles/news2018-03-14 [Zugriff 1.5.2018]
[16] Deutscher Bundestag (2018) Drucksache 19/1296. http://dip21.bundestag.de/dip21/btd/19/012/1901296.pdf
[17] BÄK (2018) Zum Vorhaben der EU-Kommission zur Zentralisierung medizinischer Nutzenbewertungen (HTA) Fachliche Einschätzung der Bundesärztekammer vom 20.04.2018 www.bundesaerztekammer.de/aerzte/qualitaetssicherung/health-technology-assessment/einschaetzung [Zugriff 1.5.2018]
Wichtige Ziele fehlen
EU Forschungsprogramm mit Mängeln
Die Ausrichtung der Europäischen Forschung läuft Gefahr, sozialen Herausforderungen nicht gerecht zu werden. Stattdessen könnten kommerzielle Interessen die Oberhand gewinnen. Das befürchten viele Organisationen der Zivilgesellschaft, nachdem die Europäische Kommission ihr Konzept für ein neues Forschungsrahmenprogramm veröffentlicht hat.
Unter dem Titel Horizon Europe sollen die europäischen Forschungsaktivitäten für den Zeitraum 2021 bis 2027 gefördert werden.[1] Die Europäische Kommission setzt dafür ein Budget von knapp 100 Milliarden € an. In einem 4-seitigen Konzeptpapier skizziert sie die Eckpunkte ihrer Strategie. Prinzipiell möchte man zwei Ziele erreichen: Wissenschaft und Technologie innerhalb der EU ausbauen sowie die globale Wettbewerbsfähigkeit der europäischen Industrie stärken.
Ein European Innovation Council soll Unternehmen dabei unterstützen, ihre Ideen zur Marktreife zu entwickeln. Auch thematische Schwerpunkte sollen in einem breiten Diskussionsprozess festgelegt werden. Als mögliche Themen nennt die Kommission den Kampf gegen Krebs, sauberen Transport oder plastikfreie Meere. Unter dem Schlagwort Open Science soll außerdem der freie Zugang zu Forschungsergebnissen konsequenter gefördert werden als es beim derzeitigen Forschungsrahmenprogramm Horizon 2020 der Fall ist: Open Access Publikationen sowie die Offenlegung von Daten sollen verpflichtend werden. Die Aktivitäten werden in drei so genannten Säulen zusammengefasst: Open Science (25,8 Mrd. €), Open Innovation (13,5 Mrd. €) und “Global Challenges und Industrial Competitiveness” (52,7 Mrd. €).
Offener Brief
Besonders die Säule “Global Challenges und Industrial Competitiveness” stößt auf starke Kritik. Wenn gesellschaftliche Herausforderungen und Stärkung der Industrie als ein gemeinsames Ziel benannt werden, verschleiert das starke Interessenkonflikte. Das kritisiert die Pharma-Kampagne gemeinsam mit vielen anderen europäischen NGOs in einem offenen Brief an die EU Kommission.[2]
Besser wäre es, eine eigenständige Budgetlinie für globale Herausforderungen festzulegen, die sich an den Zielen für nachhaltige Entwicklung (Sustainable Development Goals, SDGs) und dem Pariser Klimaabkommen orientiert. Ziel muss sein, Alternativen zum bisherigen Fokus auf Wachstum und Konsum zu entwickeln.
Ebenso vermissen die Unterzeichner des Briefs an die Europäische Kommission ein klares Bekenntnis, die öffentliche Förderung an Bedingungen zu knüpfen. Es muss sichergestellt werden, dass die Produkte aus öffentlich finanzierter Forschung auch größtmögliche Nutzung erfahren und vielen Menschen zugänglich sind (“public return on public investment”).
Mit ihrem Konzept hat die Kommission den Gestaltungsprozess für das Rahmenprogramm eröffnet, der in den kommenden Monaten in vielen parlamentarischen und außerparlamentarischen Foren geführt wird. Die BUKO Pharma-Kampagne wird sich hier weiterhin einbringen. (CW)
Artikel aus dem Pharma-Brief 4-5/2018, S. 5
[1] https://ec.europa.eu/commission/publications/research-and-innovation-including-horizon-europe-iter-and-euratom-legal-texts-and-factsheets_en
WHO öffnet Schleusen für private Einflussnahme
Judith Richter über Ursachen und Folgen
Im Mai 2016 haben die Mitgliedsstaaten der WHO eine Entscheidung getroffen, die weitreichende Konsequenzen für die Weltgesundheit hat: Als Herzstück der WHO-Reform wurde das Rahmenwerk zum Umgang mit nicht-staatlichen Akteuren (FENSA[1]) verabschiedet. Und zwar trotz zahlreicher Warnungen, dass diese mangelhaften Regeln den Einfluss von Unternehmen und philanthropischen Stiftungen auf die WHO verstärken würden.
Die Führung der WHO ignorierte wiederholte Aufforderungen von Mitgliedsstaaten, für adäquate Handlungsempfehlungen zum Umgang mit Interessenkonflikten zu sorgen. Sämtliche Warnungen, dass dem WHO-Rahmenwerk zur Beziehung mit nicht-staatlichen Akteuren ein falsches Verständnis von Interessenskonflikten zugrunde liege, wurden in den Wind geschlagen.[2] [3]
Außerdem gab es keine angemessene öffentliche Debatte über die Tatsache, dass FENSA eine neues Regelwerk für offizielle WHO-Beziehungen beinhaltet: Eine Änderung der Terminologie kombiniert mit der Einführung eines angeblich übergeordneten Prinzips der Einbindung aller Akteure (principle of inclusiveness) hat die jahrelange Lobbyarbeit von Unternehmen und großen Förderstiftungen mit einem Schlag legitimiert: Quasi automatisch wurden sie als nicht-staatliche Akteure in „offizieller Beziehung“ mit der WHO anerkannt.
Die Bill und Melinda Gates Stiftung war eine der ersten Organisationen, die von den neuen Bedingungen profitierte. Im Januar 2017 erhielt sie den Status als „non-State actor in offizieller Beziehung mit der WHO“ Kaum drei Jahre zuvor hatte ein hoher Berater der Rockefeller Stiftung die Erwartungen der großen Stiftungen unmissverständlich formuliert: „Wir wollen nicht bloß ein weiterer ‚nicht-staatlicher Akteur‘ sein […] Die UN und Regierungen müssen uns mit offenen Armen empfangen und günstige Rahmenbedingungen für Stiftungen einführen, sowohl national als auch über Grenzen hinweg.“[4]
Wirtschaftsverbände mussten länger warten, um als nicht-staatliche unternehmerische Akteure (BINSA [5]) Zugang zu den Führungsgremien der WHO zu gewinnen: 1982 hatte ein PR-Berater transnationalen Unternehmen geraten „wirksam“ die „regulatorische Stimmungslage“ bei der UN zu umgehen und „effektive Nicht-Regierungsorganisationen“ zu kreieren, die dann in allen nur möglichen UN-Organisationen offiziell Industrieinteressen vertreten könnten. Mit „Nicht-Regierungsorganisationen“ waren dabei Wirtschaftsverbände wie der Pharmaverband IFPMA gemeint. Dieser betrieb damals eine vehemente Lobbyarbeit und untergrub so die Arbeit der WHO an einem verbindlichen internationalen Arzneimittel-Kodex.[6]
1987 übernahm der Nestlé Geschäftsführer Helmut Maucher die Präsidentschaft der internationalen Handelskammer. In einem Artikel für die Financial Times unter dem Titel „Regieren durch Konsens“ sagte er: „Regierungen müssen verstehen, dass die Wirtschaft nicht nur ein weiterer Interessenverband ist, sondern eine Ressource, die ihnen helfen wird, die richtigen Regeln zu machen.“[6]
Falsche Definitionen
FENSA definiert sowohl individuelle als auch institutionelle Interessenskonflikte. Auf den ersten Blick scheinen sie den Definitionen zu ähneln, die derzeit die meisten Ärztekammern verwenden. Die meisten gehen auf eine Definition von Denis F. Thompson zurück, die das US- Institute of Medicine[7] unter dem Titel „Interessenkonflikte in der Arzneimittelforschung, Lehre und Praxis“ veröffentlichte.[8]
FENSA enthält auf den ersten Blick nur leicht veränderte Definitionen, die in Wirklichkeit aber das ganze Konzept verwässern: „Ein Interessenkonflikt entsteht in Situationen, die das Potenzial haben, sekundäre Interessen zu berühren (Eigeninteresse an einem Arbeitsergebnis der WHO in einem bestimmten Bereich) und damit die Unabhängigkeit oder Objektivität eines professionellen Urteils oder von Handlungen in Bezug auf das primäre Interesse (die Arbeit der WHO) in unangemessener Weise beeinflussen oder zumindest diesen Eindruck erwecken.“ (§ 22)
„Alle Institutionen haben vielfältige Interessen, was bedeutet, dass die WHO bei der Zusammenarbeit mit nicht-staatlichen Akteuren häufig mit einer Kombination aus konvergierenden und gegensätzlichen Interessen konfrontiert ist. Ein institutioneller Interessenkonflikt ist eine Situation, in der die primären Interessen der WHO, die sich aus ihrer Verfassung ergeben, möglicherweise durch den Interessenkonflikt eines nicht-staatlichen Akteurs unangemessen beeinflusst werden oder zumindest der Eindruck erweckt wird, dass er sich auf die Unabhängigkeit und Objektivität der Arbeit der WHO auswirkt […]“ (§ 24)
Diese Definitionen verwischen wichtige Differenzierungen. Darauf wurde schon bei der Weltgesundheitsversammlung 2014 hingewiesen als die Mitgliedsstaaten sich weigerten, den ersten Entwurf von FENSA zu verabschieden.
Bei einer WHO Fachtagung im Jahr 2015, die sich mit dem Umgang mit Interessenskonflikten bei der Planung und Umsetzung von Ernährungsprogrammen auf Länderebene auseinandersetzte, äußerten Experten sich besorgt über ein irreführendes Hintergrundpapier dieser WHO Tagung. Unter anderem vermischte es das FENSA Konzept und Thompsons Konzept zu Interessenkonflikten von 1993.
Die Diskussion zeigte, dass die allgemeinen Definitionen von Interessenkonflikten nicht präzise genug waren. Experten rieten daher „auf andere existierende Definitionen zurückzugreifen“. Die spezifischen Definitionen von Interessenkonflikten entsprächen „nicht dem Standard der Rechtspraxis“.[9]
Der vieldeutige Begriff „vested interest“ sollte nicht in Analysen von Interessenkonflikten verwendet werden, sondern durch Begriffe wie „finanzielle“ oder „persönliche Interessenkonflikte“ ersetzt werden, die innerhalb einer Person oder Organisation auftreten und nicht Konflikte alle Art zwischen verschiedenen Akteuren. Eine Unterscheidung, die von der Juraprofessorin Ann Peters in einer Analyse von 2012 vorgeschlagen wurde, die Interessenkonflikte in der globalen Governance thematisierte.[10]
Warum klammert sich die WHO an fragwürdige Definitionen?
Das fragt man sich wirklich. Hätte die Führung der WHO sich auf die relativ simple Definition institutioneller Interessenkonflikte der IoM berufen, hätte sie argumentieren können, dass die WHO ihre wesentlichen institutionellen Interessenkonflikte – verursacht durch die Abhängigkeit von „freiwilliger“ Finanzierung – nicht lösen könne, solange Mitgliedsstaaten sich weigerten, ihre seit Jahren unverändert niedrigen Beiträge anzuheben.
Anstatt auf „zahlreiche Interessen“ von „allen Institutionen“ und Kombinationen von „übereinstimmenden“ und „gegensätzlichen Interessen“ zu verweisen, hätte die WHO-Führung konstruktiven Vorschlägen folgen können, die bei der Auseinandersetzung um FENSA gemacht wurden. Dann hätte sie z.B. die Ausführungen von Ann Peters beachtet. Die Professorin empfiehlt, dass Konzepte zu Interessenkonflikten, um rechtlich aussagekräftig zu sein, auch auf Loyalitätskonflikte Bezug nehmen sollten – also auf Konflikte, die durch divergierende Rollen ausgelöst werden oder durch Akteure, die „zwei Herren dienen“, die gegensätzliche Mandate haben. Die WHO hätte auch festgestellt, dass der Rechtsprofessor Marc Rodwin bereits 1993 so ein Konzept in seinem Buch Medizin, Geld und Moral vorschlug und es seither weiter präzisiert hat.
Die zentrale Frage heute ist: Gibt es Hoffnung, dass der neue Generaldirektor der WHO problematische Konzepte in FENSA korrigieren wird? Macht er sich als ein aus Afrika stammender Generaldirektor Gedanken darüber, dass die Bevölkerung seines Kontinents möglicherweise nicht von einem Entwicklungsmuster profitieren wird, das „freiwillige“ Finanzierung und private Investitionen anpreist? Und darüber, dass im Gegenzug der Einfluss von Konzernen oder der von superreichen Unternehmen gegründeten Stiftungen ausgedehnt wird – sei es in globalen „Partnerschaften mit multiplen Interessenvertretern“ (Stakeholder) oder ganz direkt am Tisch derer, die Politik machen.
Was kann man tun?
Die Verabschiedung der FENSA-Politik hat auch den Diskurs über „Partnerschaften“ neu belebt, die schon in der frühen Phase der WHO Reform kritisch hinterfragt wurden. 2011 schenkten die Mitgliedsstaaten der WHO der Zivilgesellschaft Gehör, als sie Kritik an den Begriffen „Interessenvertreter“ und „Partnerschaft“ äußerte, weil diese Terminologie grundlegende Unterschiede zwischen den Akteuren verschleiert. Damals weigerten sich die Mitgliedsstaaten, ein „Multi-Stakeholder“ Weltgesundheitsforum zu gründen, dem die Generaldirektorin Dr. Margaret Chan bereits zugestimmt hatte. Sie forderten Schutzmaßnahmen, die klar unterscheiden sollten zwischen Akteuren aus dem privaten Sektor und anderen Akteuren.
FENSA ist das Ergebnis dieser Debatte. Es birgt die Gefahr, dass es den Weg ebnet für eine Zunahme schädlicher Verstrickungen, anstatt für ein angemessenes Verhältnis zwischen der WHO, Akteuren aus der Wirtschaft (BINSAs) und Stiftungen zu sorgen.
Eine Überprüfung von FENSA ist vorgesehen. Aber sie wird viel zu spät kommen, um noch verhindern zu können, dass das Konzept von Interessenkonflikten bzw. Strategien zu deren weltweiter Regulierung ausgehöhlt werden.
SOS
Das Schiff der globalen Gesundheit wird mehr denn je gelenkt von denen, die jetzt auf ihr Recht auf „Einbindung“ als besonders wertvolle „Stakeholder“ pochen können. Sagen Sie „Nein“ zur „Stakeholderisierung“ von öffentlichen Foren und Diskursen und drängen sie auf sofortige Korrektur der WHO-Konzepte zu Interessenskonflikten.
Judith Richter ist Sozialwissenschaftlerin (PhD Soc.) und Apothekerin und forscht zu Regulierung von multinationalen Firmen und demokratischer Regierungsführung. Eine englische Vorversion wurde auf der Mezis-Tagung am 15.9.2017 in Berlin als Poster präsentiert.
https://mezis.de/wp-content/uploads/2017/09/JR_2017_09_Mayday_WHO_CoI-FINAL.pdf
Übersetzung: Antonia Wellmann
Artikel aus dem Pharma-Brief 1/2018, S. 1
[1] FENSA: Framework for engagement with non-state actors www.who.int/about/collaborations/non-state-actors/en/
[2] Richter J (2014) Time to debate WHO’s understanding of conflict of interests. BMJ www.bmj.com/content/348/bmj.g3351/rr
[3] Richter J (2017) Comments on Draft Approach for the prevention and management of conflicts of interest in the policy development and implementation of nutrition programmes at country level. WHO online consultation, 29 Oct www.who.int/nutrition/consultation-doi/judith_richter.pdf
[4] Martens J and Seitz K (2015) Philanthropic power and development. Who shapes the agenda? Aachen/Berlin/Bonn/New York: Brot für die Welt u.a.
[5] BINSA: Business interest non-state actor. Dieser von Judith Richter geprägte Begriff soll der besseren Unterscheidung der unterschiedlichen Interessen von kommerziellen nichtstaatlichen Akteuren und denjenigen, die das Allgemeinwohl vertreten (PINSA: Public interest non-state actor), deutlich machen.
[6] Zitiert in: Richter J (2001) Holding corporations accountable. London: Zed Books
[7] Seit 2015 National Academy of Medicine (NAM)
[8] http://nationalacademies.org/hmd/activities/workforce/conflictofinterest.aspx
[9] WHO (2016) Addressing and managing conflicts of interest in the planning and delivery of nutrition programmes at country level. www.who.int/nutrition/publications/COI-report/en/
[10] Siehe Fußnote 9, p 4-6
Wenn Autoren lügen
Doppelveröffentlichungen verfälschen Wissenschaft
Wenn Autoren dieselbe Studie zweimal veröffentlichen, aber den Eindruck erwecken es handele sich um verschiedene Forschungsarbeiten, führt das zu falschen Schlussfolgerungen über den Nutzen der untersuchten Arzneimittel. Drei spanische Autoren kamen einem größeren Betrug auf die Spur.[1]
Luis Carlos Saiz und Kollegen aus Pamplona starteten 2015 eine systematische Übersichtsarbeit zu Blutdrucksenkern. Dabei fiel ihnen auf, dass acht klinische Studien, die alle denselben Hauptautor hatten, große Ähnlichkeiten aufwiesen. Nachfragen beim Autor verstärkten den Verdacht, dass gemogelt worden war. Um Klarheit zu bekommen, fragten die drei bei den Herausgebern der Zeitschriften nach. Schließlich kam heraus, dass es sich in Wirklichkeit nicht um acht, sondern nur um zwei Studien handelte. Eine schwerwiegende Angelegenheit, weil in allen Artikeln ein Vorteil für das neue Medikament reklamiert worden war. Immerhin hatte die investigative Arbeit von Saiz und Kollegen ein Nachspiel: Außer den beiden Erstveröffentlichungen wurden alle anderen Artikel inzwischen von den Zeitschriften zurückgezogen.
Wer einmal lügt …
Das International Committee of Medical Journal Editors (ICMJE) warnt, dass „die Gültigkeit früherer Veröffentlichungen eines Autors, der des Betruges überführt wurde, nicht mehr vorausgesetzt werden kann.“ Diese Annahme veranlasste die drei Spanier, andere Publikationen unter die Lupe zu nehmen. Sie fanden weitere 121 Veröffentlichungen desselben Verfassers. Bei 78 Artikeln war er der Hauptautor, davon waren die Hälfte (39) der Doppelveröffentlichung verdächtig. Dagegen waren die 53 Veröffentlichungen, bei denen er nur Mitautor war, unverdächtig.
Tarnen und täuschen
Kriterium für die Identifizierung der Dubletten war eine große Zahl identischer Daten in den verschiedenen Artikeln. Hinter den 39 Veröffentlichungen steckten nach Analyse von Saiz und Kollegen nur 15 klinische Studien. Neun waren zweimal, drei dreimal oder gar viermal unter verschiedenen Überschriften veröffentlicht worden.
Auch sonst wurde allerlei unternommen, um alten Wein in neue Schläuche zu gießen. Tabellen bekamen andere Titel und die grafischen Darstellungen der Ergebnisse waren unterschiedlich gestaltet. Meist vermied es der Autor, die anderen Veröffentlichungen zur selben Studie zu zitieren. Nirgends war ersichtlich, dass er dieselben Forschungsergebnisse mehrfach publiziert hatte.
Im Oktober 2015 schrieben Saiz und Kollegen die Herausgeber der 22 betroffenen Zeitschriften an. Die meisten reagierten zwar prompt mit einer Antwort. Zwei Drittel (64%) der Zeitschriften hatten jedoch auch zwei Jahre später noch keine endgültige Entscheidung über die inkriminierten Artikel getroffen. Nur vier Artikel wurden zurückgezogen. Fünf Veröffentlichungen wurden als Originalarbeiten identifiziert und fünf weitere hielten die Herausgeber für unterschiedlich genug, um nicht als Duplikat zu gelten. Bei vier anderen Artikeln wird noch diskutiert, ob sie zurückgezogen oder korrigiert werden. Nach zwei Jahren bleiben also immer noch 21 zweifelhafte Studienpublikationen übrig, wo der Ausgang völlig offen ist.
Die Wissenschaftler aus Pamplona kritisieren, dass es zwar vom ICJME, dem Committee on Publication Ethics (COPE) und dem Council of Science Editors (CSE) klare Regeln zum Umgang mit gefälschten Publikationen gibt, aber keine Fristen existieren. Das führt dazu, dass irreführende Artikel noch jahrelang den wissenschaftlichen Diskurs beeinflussen. Die meisten der fraglichen Veröffentlichungen wurden mehr als zehnmal in anderen Artikeln zitiert, einige über 50-mal. Die spanischen Autoren fordern deshalb auch klare Fristen zum Rückzug von offensichtlichen Fälschungen.
Alle beanstandeten Veröffentlichungen hatten ein Thema: Diabetes. Gerade auf diesem Forschungsgebiet zeigt sich eine hohe Konzentration auf wenige „Vielschreiber“. Holleman und KollegInnen identifizierten über einen Zeitraum von 20 Jahren 991 klinische Studien zu blutzuckersenkenden Medikamenten.[2] An den 3.782 Veröffentlichungen zu diesen Studien wirkten insgesamt 13.592 AutorInnen mit. Aber gerade einmal 110 schrieben an einem Drittel der Veröffentlichungen mit. Davon waren 44% Firmenangestellte und 56% WissenschaftlerInnen, die alle enge Beziehungen zur Pharmaindustrie pflegten.
Bleibt die beunruhigende Frage, ob AutorInnen, die eine Mehrfachveröffentlichung derselben Studie verschleiern, nicht vielleicht auch an anderen Stellen schummeln. Deshalb wäre die verpflichtende Veröffentlichung der Clinical Study Reports (CSR), die die vollständigen Ergebnisse enthalten, auch so wichtig. Die entsprechende EU-Verordnung sieht das für neu zugelassene Arzneimittel auch vor. Allerdings geschieht die Umsetzung zögerlich. Und ältere Studien bleiben eine Black Box, obwohl sie eine wichtige Basis für die ärztlichen Behandlungsempfehlungen sind. (JS)
Artikel aus dem Pharma-Brief 4-5/2018, S. 3
[1] Saiz LC et al (2018) When authors lie, readers cry and editors sigh. BMJ Evidence Based Medicine; 23, p 92
[2] Holleman F et al. (2015) Productivity of authors in the field of diabetes. BMJ; 350, p h2638
Welches Wachstum wollen wir?
Was ist Wohlstand? Und wie messen wir den Wert, den unsere Wirtschaft hervorbringt? Für die britische Ökonomin Mariana Mazzucato sind das grundlegende Fragen für die Gerechtigkeit in der Gesellschaft. In ihrem neusten Buch fordert sie deshalb eine Debatte über unser Verständnis von Wirtschaftswachstum – auch am Beispiel der Pharmaindustrie.
Das Buch beginnt mit einem Gang durch die Geschichte der Wirtschaftswissenschaften. Die Autorin verdeutlicht dabei, wie die Wirtschaftstheoretiker sich über Jahrhunderte mit der Frage auseinandersetzen, was denn eigentlich „Wert“ bedeutet und was „Wohlstand“ schafft.
Sie beginnt mit der im 16. Jahrhundert entstandenen Theorie des Merkantilismus, der den Handel in den Mittelpunkt stellte, über die Physiokratie mit Grund und Boden als Quelle des Reichtums, bis zur klassischen Ökonomie mit dem Fokus auf Arbeitskraft.
Casino-Kapitalismus
Aktuell wichtige Messgröße für die Stärke eines Wirtschaftsraums ist das Bruttoinlandsprodukt (BIP). Auch hier hat sich die Definition im Lauf der letzten Jahrzehnte verändert. Erst in den 1970er Jahren wurde der Finanzsektor in die Berechnung des Bruttoinlandsprodukts (BIP) einbezogen. Mit Aufkommen des Casino-Kapitalismus verschwammen die Grenzen zwischen Handelsbanken (primäre Aufgabe: Geld bereitstellen) und Investmentbanken (primäres Ziel: Gewinnmaximierung). Gleichzeitig begann die Deregulierung der globalen Finanzmärkte. Infolge hat sich in den USA zwischen 1975 und 2015 das BIP verdreifacht und die Produktivität ist um 60% gestiegen. Die Reallöhne dagegen stagnierten oder sanken sogar.
Gleichzeitig hat sich die Schere zwischen arm und reich global noch weiter geöffnet: Der Besitz der 62 reichsten Menschen ist um 45% gewachsen, das Vermögen der unteren Hälfte der Weltbevölkerung um 38% gesunken.
Nach der heute gängigen Definition des BIP hat zwar die moderne Finanzindustrie enorme Werte geschaffen, gesamtgesellschaftlich jedoch keinen Wohlstand erzeugt. Das festzuhalten ist Mazzucato wichtig, denn „Wirtschaftswissenschaft ist im Kern eine Sozialwissenschaft“.
Pharmaindustrie
Neben der Finanzindustrie unterzieht die Autorin auch die Pharmaindustrie einer kritischen Prüfung. Aufhänger ist das Hepatitis-Medikament Harvoni ® (Sofosbuvir). Der Anbieter Gilead wurde wegen der hohen Preise stark kritisiert. Die Firma argumentiert mit einem “value-based pricing”: Das Medikament rette Leben und helfe, an anderer Stelle Kosten einzusparen. Dieser Wert rechtfertige den hohen Preis.
Mazzucato widerlegt diese Argumentation damit, dass nachgewiesenermaßen bei Medikamenten kein Zusammenhang zwischen Preis und therapeutischem Nutzen besteht. Zudem macht sie deutlich, dass Pharmaunternehmen das Konzept des value-based pricing ins Gegenteil verkehrt haben – es wurde nämlich ursprünglich in Großbritannien am NICE dazu entwickelt, das öffentliche Gesundheitsbudget sinnvoll einzusetzen und Kosten zu sparen.
Ebenso interessant sind Parallelen zur Entwicklung der Finanzindustrie. Die moderne biopharmazeutische Industrie geht auf umfangreiche Anschubfinanzierung der Grundlagenforschung durch die staatlichen US-amerikanischen National Institutes of Health zurück. Die darauf folgenden Firmengründungen gehen einher mit der Entstehung einer Risikokapital-Industrie, die enorme Börsenwerte erzeugt hat, obwohl nur wenige Biopharma-Firmen wirklich erfolgreich Produkte auf den Markt bringen konnten. Mazuccato sieht auch hier den Finanzmarkt als Hauptgewinner.
Staat muss steuern
Aus ihrer umfangreichen Analyse verschiedener Industriezweige leitet die Ökonomin verschiedene Maßnahmen ab, wie staatliches Eingreifen aussehen kann, etwa eine Finanztransaktionssteuer oder Beschränkungen beim Aktienrückkauf als Mittel der Steuervermeidung.
Diskussionswürdig ist der Vorschlag, staatliche Investmentbanken zu gründen, die sich klar definierten Zielen widmen (“mission oriented”). Als Vorbild nennt Mazzucato die Mond-Mission, die nicht das Ziel hatte, einen bestimmten Wirtschaftssektor zu unterstützen, sondern eben das Ziel, den Mond zu erreichen. Solche Ziele z.B. im Bereich Gesundheit könnten von öffentlichen und privaten Akteuren gemeinsam erreicht werden mit Hilfe von zielgerichteten, langfristigen Risikoübernahmen – unter der Bedingung, dass beispielsweise Arzneimittelpreise die Risikoverteilung zwischen privat und öffentlich reflektieren. Mazzucato will mit ihrem Buch eine Debatte über die Frage anstoßen, welche Art von Wachstum wir wollen. Dabei solle es nicht um die Wachstumsrate gehen, sondern um die Wachstumsrichtung. Dazu kann dieses anschaulich geschriebene Buch einen wichtigen Beitrag leisten – auch wenn ihre Lösungsansätze wie beim Beispiel Pharma noch nicht konsequent ausgearbeitet sind.
Artikel aus dem Pharma-Brief 4-5/2018, S. 6
Bild: Cover von Mazzucato M (2018) The value of everything. Making and taking in the global economy. London: Allen Lane, 364 S., 15,99 £
Von wegen Durchbruch
Analyse von “Breakthrough Therapies” in den USA ernüchternd
Mit dem “Breakthrough Therapy Program” hatten die USA 2012 das vierte Programm für eine beschleunigte Arzneimittelzulassung verabschiedet. Aber stellen die auf diesem Weg zugelassenen Medikamente wirklich einen therapeutischen Fortschritt dar? Dieser Frage gingen drei Wissenschaftler der Yale University in den USA nach.[1]
Von 2013 bis 2017 wurden 46 Medikamente als “Breakthrough Therapy” zugelassen – als Therapien also, denen ein bedeutender medizinischer Durchbruch bescheinigt wird. Das sind viel mehr als erwartet. Die US-Behörde FDA hatte mit zwei solcher Zulassungen pro Jahr gerechnet.
Für die 46 Wirkstoffe wurden 89 klinische Studien eingereicht. Über die Hälfte der Zulassungen stützt sich also auf nur eine einzige Arzneimittelstudie. Und auch die Qualität der Studien lässt oft zu wünschen übrig. Denn gemessen wurden meist (78,3%) nur Surrogatendpunkte, also keine für die PatientInnen relevanten Ergebnisse wie eine längere Überlebensdauer, die Reduzierung von Krankheitssymptomen oder eine Verbesserung der Lebensqualität.
Datenlage mau
Für 41,3% der Medikamente gab es keine randomisierten Studien, weniger als die Hälfte waren verblindet. Beides ist eigentlich wissenschaftlicher Standard, um Verzerrungen zu vermeiden. Zum Teil wurden Vergleichsdaten aus älteren Studien herangezogen (historischer Vergleich) oder es wurde mit gar nichts verglichen. Letzteres war bei den 25 Krebsmedikamenten, die als potenzieller Durchbruch eingestuft wurden, auffällig häufig der Fall: Nur 10 dieser Mittel wurden gegen ein anderes Medikament oder gegen Placebo getestet.
Auch die PatientInnenzahl war in vielen Studien gering. Das betraf – wenig überraschend – besonders die Orphan Drugs gegen seltene Erkrankungen, die 30 der 46 Breakthrough Therapies ausmachten.
Evidenz unbefriedigend
Was die Sache noch schlimmer macht: Die Evidenz bleibt auch Jahre später noch immer unbefriedigend. Entweder weil bessere Daten gar nicht erst generiert werden, oder weil weitere Studien ergaben, dass sich die ursprünglichen Hoffnungen nicht erfüllen. Kim und Prasad hatten 2015 gezeigt, dass bei Krebsmedikamenten, die von 2008 bis 2012 auf Basis von Surrogaten zugelassen worden waren, vier Jahre später[2] bei der Hälfte klar war, dass sie keinen Überlebensvorteil bieten. Nur bei 14% der Medikamente bestätigten sich die Erwartungen und bei 36% war der Nutzen immer noch unklar.[3]
Bereits 2014 übten Wissenschaftler scharfe Kritik an dem Breakthrough Therapy Program.[4] Denn nach dem Start im Jahr 2013 wurden innerhalb von nur neun Monaten 27 Medikamente in das Beschleunigungsprogramm aufgenommen. Angesichts des bis dato üblichen Schnitts von jährlich 25 Zulassungen sei das aber „kein Hinweis auf eine plötzliche und dramatische Zunahme im Tempo der pharmazeutischen Innovation“, so Darrow und Kollegen. „Eine andere Interpretation für die schnelle Popularität dieses Programms ist, dass es den Anschein von Fortschritt erzeugt und gleichzeitig die Sichtbarkeit von vielversprechenden Produkten im frühen Entwicklungsstadium erhöht. Diese Arzneimittel bieten PatientInnen aber nicht häufiger einen großen Nutzen als das vor der Einführung des Gesetzes der Fall war. Die Zuordnung zur Breakthrough Therapy wird wahrscheinlich auch den öffentlichen Druck auf die FDA erhöhen, solche Produkte am Ende zuzulassen.“ (JS)
USA Beschleunigungsverfahren [5]
Accelerated approval (1992) Durch die Verwendung von Surrogatindikatoren statt harten klinischen Endpunkten können Studien schneller abgeschlossen und so die Zulassung beschleunigt werden.
Priority review (1992) Zulassungsentscheidung innerhalb von 6 statt 10 Monaten
Fast-track (1997) und Breakthrough therapy program (2012) erlauben eine kürzere Studiendauer. Das zweite Programm beinhaltet zusätzlich eine frühe Beratung des Herstellers durch leitende FDA-MitarbeiterInnen und intensive Unterstützung während des gesamten Zulassungsprozesses.
Artikel aus dem Pharma-Brief 8-9/2018, S.5
[1] Puthumana J et al. (2018) Clinical Trial Evidence Supporting FDA Approval of Drugs Granted Breakthrough Therapy Designation. JAMA; 320, p 301
[2] Im Median 4,4 Jahre nach Zulassung
[3] Kim C and Prasad V (2015) JAMA Int Med; 175, p 1992
[4] Darrow JJ et al. (2014) New FDA Breakthrough-Drug Category – Implications for Patients. N Engl J Med 370; p 1252
[5] Hwang TJ et al. (2017) The FDA’s Expedited Programs and Clinical Development Times for Novel Therapeutics, 2012-2016. JAMA; 318, p 2137
Verschleppte Warnungen
Frühe Warnzeichen bei Schweinegrippeimpfstoff ignoriert
Als 2009 weltweit die Schweinegrippe grassierte, empfahl die Weltgesundheitsorganisation Massenimpfungen. Die Entscheidung war stark umstritten, denn es hatte sich früh gezeigt, dass die Grippeepidemie damals eher milder verlief als in anderen Jahren.[1] Der Impfstoff Pandemrix®, der in vielen Ländern verwendet wurde, löste zudem bei manchen Menschen heftige Gegenreaktionen aus. Durch ein Gerichtsverfahren sind jetzt Dokumente öffentlich geworden, die kein gutes Licht auf den Umgang mit Risiken durch Hersteller und Behörden werfen.[2]
Die Bundesregierung hatte 2009 50 Millionen Dosen Pandemrix® geordert.[3] Der Impfstoff enthielt den Wirkverstärker AS03, dadurch konnte laut Hersteller GSK mit weniger Grippe-Antigen eine ähnliche Wirksamkeit erzeugt werden wie bei traditionell hergestellten Impfstoffen, die nur das Antigen enthalten. Der Vorteil: Der Impfstoff konnte wesentlich schneller hergestellt werden, denn die Produktion von Antigenen ist zeitraubend. Der Haken an der Sache: Die Verträglichkeit des Wirkverstärkers war nicht gut untersucht. Es gab schon früh den Verdacht, dass er überschießende Immunreaktionen auslösen könnte.[4] Auch Narkolepsie, also unkontrollierbare plötzliche Schlafattacken, wurden mit Pandemrix® in Verbindung gebracht.
Vorgeschichte
Schweden kam bereits 2010 zu dem Schluss, dass Pandemrix® in seltenen Fällen eine Narkolepsie auslösen kann und nahm die Impfempfehlung für diesen Wirkstoff zurück.[5] Basis war eine Untersuchung der Zulassungsbehörde, die sich auf Meldungen zu unerwünschten Wirkungen stützte.[6] Eine nachträgliche Analyse in Finnland ergab für Kinder zwischen 3 und 18 Jahren ein 12,7-fach erhöhtes Risiko an Narkolepsie zu erkranken, wenn sie mit Pandemrix® geimpft waren. Pro 16.000 Kindern trat ein zusätzlicher Fall an Narkolepsie auf.[7] Eine 2013 in England durchgeführte Analyse kam zu ähnlichen Ergebnissen. Beide Studien beschränkten sich auf den Zeitraum bis Ende 2010, also bevor die Medien in den Ländern über einen möglichen Zusammenhang zwischen dem Impfstoff und Narkolepsie berichteten.
Im Zuge der Impfwelle 2009/2010 hatten die Behörden in Deutschland und England die Öffentlichkeit immer wieder beschwichtigt: Der Impfstoff mit Wirkverstärker habe keine höheren Risiken und sei gründlich getestet.[8],[2] Das entsprach aber nicht den Tatsachen. Weder war der Wirkverstärker ausreichend geprüft worden noch die tatsächlich verwendeten H1N1-Antigene. Die Zulassung beruhte auf einem nie eingesetzten Impfstoff gegen die Vogelgrippe. Die Antigene waren ohne weitere Überprüfung einfach ausgetauscht worden. Der Vogelgrippe-Impfstoff war an wenigen Gesunden getestet worden und dabei hatte sich gezeigt, dass der Impfstoff mit Wirkverstärker mehr unerwünschte Ereignisse auslöste als ein Produkt ohne diesen Zusatzstoff.[9]
Opfer klagt
Eine Frau aus Irland, die nach Pandemrix®-Impfung 2009 an Narkolepsie erkrankte, verklagte den Staat und den Hersteller GSK. Letztes Jahr erhielt das Gericht und damit auch die Klägerin die Sicherheitsberichte zum Impfstoff. Die Dokumente waren zwischen Dezember 2009 und März 2010 innerhalb der Firma im Umlauf und sind zumindest an die irische Kontrollbehörde geschickt worden. Der von der Klägerin hinzugezogene Experte, der angesehene Mediziner und Epidemiologe Tom Jefferson, sagte: „Als ich die Tabellen sah, bin ich beinah vom Stuhl gefallen. Jeder Verbraucher kann erkennen, was hier los ist.“[2] „Das Erstaunliche war, dass niemand die Daten ausgewertet hatte“, so Jefferson, obwohl die Zahl der unerwünschten Wirkungen und der verwendeten Impfdosen bekannt war.
GSK hatte bis Ende November 2009 bereits 1.138 Berichte über schwere unerwünschte Wirkungen (UAW) zu Pandemrix® erhalten. Bis März 2010 war die Zahl auf 5.069 angestiegen, das entspricht 72 schweren UAW pro einer Million Impfdosen. Das eigentlich Überraschende ist aber, dass das in einem anderen Land von GSK für andere Märkte hergestellte Arepanrix®, das ebenfalls den Wirkverstärker enthielt, deutlich seltener mit unerwünschten Wirkungen in Verbindung gebracht wurde: Schwere UAW waren bei Pandemrix® fünfmal so häufig. Die Zahlen von GSK sind allerdings insofern mit etwas Vorsicht zu genießen, weil GSK die Werte von Arepanrix® und einem Impfstoff ohne Wirkverstärker zusammengeworfen hat.
Das ändert nichts daran, dass bei der Firma und den Zulassungsbehörden die Alarmglocken hätten schrillen müssen, denn offensichtlich war Arepanrix® und erst recht der Impfstoff ohne Wirkverstärker viel besser verträglich. Eine mögliche Teilerklärung ist, dass bei dem in Dresden produzierten Pandemrix® möglicherweise bei der Produktion etwas schief gegangen ist. Nachfragen des BMJ beantwortete GSK mit Hinweis auf das laufende Gerichtsverfahren nicht.
Eine spannende Frage ist, warum weder die europäische Zulassungsbehörde EMA noch nationale Behörden etwas unternahmen (mit Ausnahme von Schweden), obwohl die Hersteller verpflichtet sind, Berichte über unerwünschte Wirkungen zeitnah mitzuteilen. Die europäische Behörde beschied dem BMJ: „Die EMA führt keine vergleichenden Nutzen-Risiko Bewertungen zwischen in der EU zugelassenen Produkten oder mit Produkten, die außerhalb der EU genehmigt sind, durch.“[2] Auf diese Aussage reagierte Tom Jefferson mit Unverständnis: „Was ist der Sinn von Pharmakovigilanz, wenn niemand etwas mit der Information anfängt? Es brauchte acht Jahre, dass diese Information durch wissenschaftliche Recherchen und Schadensersatzprozesse ans Licht kam. Ist das akzeptabel? Dass wir nur über einen Teil der Informationen verfügen, ist das unmittelbare Ergebnis von Geheimnistuerei, die keine gesundheitspolitische Maßnahme umgeben sollte.“
Peter Doshi, Autor des Berichts im BMJ, fragt am Schluss. „Welche unerwünschten Wirkungen sie auch immer verursacht haben, sie sind Impfstoffe der Vergangenheit. Aber die Ereignisse 2009-2010 werfen grundlegende Fragen zur Transparenz von Informationen auf. Wann haben die Gesundheitsbehörden die Pflicht, vor unerwünschten Wirkungen von Impfstoffen zu warnen, die durch Pharmakovigilanz entdeckt wurden? Mit wie vielen Details sollte die Öffentlichkeit versorgt werden und sollte das aktiv oder nur auf Nachfrage erfolgen? Wenn sich Geschichte wiederholen sollte, hat die Öffentlichkeit ein Recht auf Wissen?“ (JS)
Die Zeche berappt der Steuerzahler
Auch kostenmäßig war Pandemrix ® eine schlechte Wahl, denn für den Wirkverstärker berechnete die Firma einen höheren Preis als für das eigentlich Antigen. In Deutschland waren nach Berechnungen des arznei-telegramms die Kosten für den Grippeimpfstoff 150 Mio. € höher als für einen Impfstoff ohne Wirkverstärker.[2] Ganz zu schweigen davon, dass am Ende große Mengen des Impfstoffs entsorgt werden mussten, denn während die Schweinegrippe grassierte, ließen sich nur Wenige impfen. Später wurde der Impfstoff nicht mehr benötigt, weil inzwischen andere Grippeviren im Umlauf waren. 2011 war außerdem die Haltbarkeit abgelaufen. Von den 34 Mio. Impfungen, die die Bundesrepublik eingekauft hatte, wurden 29 Millionen im Wert von einer Viertelmilliarde Euro vernichtet.[10]
Artikel aus dem Pharma-Brief 8-9/2018, S.3
[1] Pharma-Brief (2010) Grippe: Vorwürfe gegen die WHO. Nr. 1, S. 8
[2] Doshi P (2018) Pandemrix vaccine: why was the public not told of early warning signs? BMJ; 362, p k3948
[3] arznei-telegramm (2009) H1N1: Fehleinschätzungen, Haftungsfreistellung und viel Geld; 40, S. 85
[4] arznei-telegramm (2009) Schweinegrippe: Alles im Griff?; 40, S. 77
[5] Miller E et al. (2013) Risk of narcolepsy in children and young people receiving AS03 adjuvanted pandemic A/H1N1 2009influenza vaccine: retrospective analysis. BMJ; 346, p f794
[6] Medical Products Agency (2011) Occurrence of narcolepsy with cataplexy among children and adolescents in relation to the H1N1 pandemic and Pandemrix vaccinations. Results of a case inventory study by the MPA in Sweden 2009-2010. www.lakemedelsverket.se/upload/nyheter/2011/Fallinventeringsrapport_pandermrix_110630.pdf
[7] Nohynek H et al. (2012) AS03 Adjuvanted AH1N1 Vaccine Associated with an Abrupt Increase in the Incidence of Childhood Narcolepsy in Finland. doi.org/10.1371/journal.pone.0033536
[8] Hackenbroch V und Traufetter G (2009) Immun gegen die Impfung. Der Spiegel online. 19. Okt. www.spiegel.de/spiegel/a-655762-3.html
[9] arznei-telegramm (2009) Schweinegrippe: Alles im Griff? ; 40, S. 77
[10] arznei-telegramm (2011) Schweinegrippeimpfstoffe für eine viertel Milliarde Euro in den Müll; 42, S. 71
Ungesunder Handel
EU verhandelt mit Lateinamerika
Gleich drei internationale Abkommen sind derzeit in Arbeit: Eines mit Mercosur, dem gemeinsamen Markt Südamerikas (Argentinien, Brasilien, Paraguay und Uruguay), sowie Neuverhandlungen von bestehenden Verträgen mit Mexiko und Chile. Die European Public Health Alliance (EPHA) hat die gesundheitlichen Aspekte der Entwürfe unter die Lupe genommen.[1]
Nachdem die Verhandlungen zum transatlantischen Handelsvertrag TTIP zwischen der EU und den USA eingefroren sind, versucht die EU-Kommission, günstige Bedingungen für Handel und Industrie in anderen Weltregionen auszuhandeln.
Die Generaldirektion für Gesundheit und Nahrungsmittelsicherheit der EU (DG SANTE) hat 2016 einen strategischen Plan für die nächsten vier Jahre erarbeitet, der auch schon für die TTIP-Verhandlungen galt. Erstes Ziel: „Ein neuer Schub für Jobs, Wachstum und Investitionen in der EU.“ [2]
Diese wirtschaftsfreundliche Haltung spiegelt sich auch in den Positionen der EU in den aktuellen Verhandlungen mit Lateinamerika wider. Eines der Hauptziele ist eine Senkung von Zöllen für Nahrungsmittel, Tabak und Medikamente. Ein anderes der Schutz von Direktinvestitionen europäischer Firmen in diesen Ländern.
Tabakförderung?
Ein weiterer Erfolgsindikator sind Direktinvestitionen im Nahrungs- und Pharmasektor von EU-Mitgliedsstaaten im Ausland. 2016 schrieb DG SANTE (noch mit Blick auf TTIP) dazu: „Bedauerlicherweise enthalten die Zahlen zum Ernährungssektor auch Tabak und Getränke und sie können aus Vertraulichkeitsgründen nicht getrennt dargestellt werden.“ [1]
Tabakkonsum kostet nach Schätzungen der Kommission jährlich 700.000 Menschen in der EU das Leben. In allen Mercosur-Staaten, Chile und auch Mexiko zählt Rauchen zu den Top fünf der Verursacher von verlorenen gesunden Lebensjahren.
Sollte also auch für die Tabakindustrie Investitionsschutz vereinbart werden, würden damit zugleich regulative Maßnahmen zum Schutz von VerbraucherInnen vereitelt – etwa neutrale Verpackungen für Zigaretten. Uruguay hat in diesem Zusammenhang bereits unangenehme Erfahrungen gemacht. Aufgrund des Investitionsschutzabkommens zwischen der Schweiz und Uruguay wurde das Land von Philip Morris vor ein Schiedsgericht gezerrt. Letztlich verlor die Zigarettenfirma zwar, aber die beträchtlichen Prozesskosten wurden Uruguay erst nach sechs Jahren erstattet.
Ungesundes Essen
Alle drei Vertragsentwürfe der EU beschäftigen sich mit der Beseitigung von Handelshemmnissen, die durch unterschiedliche Standards für Lebensmittel entstehen. Davon wären zum Beispiel die Warnhinweise auf Produkten mit hohem Kalorien-, Zucker- oder Salzgehalt in Chile betroffen. Der Abbau von Handelsschranken ist hier kontraproduktiv und untergräbt den Verbraucherschutz. Bereits heute sind in lateinamerikanischen Ländern viele Menschen stark übergewichtig und Diabetes sowie Herz-Kreislauf-Erkrankungen nehmen rasch zu. Schon 2015 stellte die PAHO fest, dass es in der Region einen deutlichen Zusammenhang zwischen dem Vordringen von stark verarbeiteten kalorienreichen Lebensmitteln, Zuckerbrausen und Übergewicht und Diabetes gibt.[3] Bislang spielte dabei vor allem das Vordringen US-amerikanischer Firmen und ihrer ungesunden Produkte die größte Rolle.
Aber auch für Europa könnte die Liberalisierung des Ernährungssektors negative Folgen haben: Zucker würde deutlich billiger werden. Auch Fleischimporte nähmen zu. Sinkende Preise würden den Konsum erhöhen mit ebenfalls negativen gesundheitlichen Folgen. Ein zusätzliches Problem dabei: eine größere Fleischproduktion geht zu Lasten der Versorgung der Menschen in Lateinamerika mit Getreide, Mais, Obst und Gemüse. Die bereits jetzt oft zweifelhaften Produktionsbedingungen von Fleisch in der EU würden sich weiter verschlechtern.
Alkoholhaltige Getränke könnten ebenfalls zu den von Handelshemmnissen befreiten Produkten gehören. Die negativen Folgen für die Gesundheit sind hinreichend bekannt.
Zugang zu Medikamenten
Im Dezember 2017 sickerten Dokumente zu den Mercosur-Verhandlungen durch, die eine ambivalente Haltung der EU zu geistigen Eigentumsrechten zeigen. So werden Datenexklusivität und zusätzliche Schutzzertifikate erwähnt, die faktisch den Patentschutz verlängern. Beides verzögert die Einführung von Generika.
Ob es sich um Tabak, ungesunde Ernährung oder teure Medikamente handelt, alle diese Probleme müsste die EU eigentlich angehen, denn sie hat sich den nachhaltigen Entwicklungszielen der UN verpflichtet (Sustainable Development Goals, SDGs). Das gilt für die Politik in den Mitgliedsstaaten ebenso wie für die Außenpolitik. Schon deshalb verbietet sich eine Wirtschaftspolitik, die der Gesundheit schadet. (JS)
Artikel aus dem Pharma-Brief 4-5/2018, S. 4
[1] EPHA (2018) Unhealthy trades. The side-effects of the European Union’s Latin American trade agreements. Brussels. https://epha.org/unhealthy-trades-the-side-effects-of-the-european-unions-latin-american-trade-agreements-report
[2] EU (2016) DG Health & Food Safety. Strategic Plan 2016-2020. https://ec.europa.eu/info/sites/info/files/strategic-plan-2016-2020-dg-sante_may2016_en_1.pdf
[3] PAHO (2015) Ultra-processed food and drink products in Latin America: Trends, impact on obesity, policy implications. Washington, DC
Theater gegen Klimawandel
Schluck & weg auf Deutschlandtour
Vom 9.-23.9. trat die Straßentheatergruppe der Pharma-Kampagne in 13 Städten quer durch die Republik auf. Für alle, die nicht dabei waren, hier ein paar Eindrücke vom Stück.
Im Königreich von Prinzessin Isabella Diarrhoea grassieren schreckliche Krankheiten. Sie versucht den Schuldigen auf die Spur zu kommen. Prinz von der Ecke versagt als erster kläglich.

Die Palastwache ist dem Ansturm der Malaria-Moskitos, die sich wegen des Klimawandels vermehren, nicht gewachsen.

Drei junge Heldinnen machen sich auf die Suche nach Opfern und den Übeltätern.

Der Bäuerin vertrocknen die Pflanzen, denn der Regen bleibt aus.
[…]

Am Ende sind sich alle einig: Wir müssen uns entschieden gegen den Klimawandel engagieren. Die Straßentheatergruppe machte damit Ernst und schob spontan einen Auftritt im Hambacher Forst ein. Der Wald soll dem Braunkohleabbau weichen.
Bilder Schluck & weg © Jörg Schaaber und Claudia Jenkes
Artikel aus dem Pharma-Brief 7/2018, S.7
Studien in Peru
Aufklärung der PatientInnen mangelhaft
Immer mehr klinische Studien werden in Entwicklungsländern durchgeführt, doch über die Bedingungen ist wenig bekannt. Deshalb ist eine aktuelle Untersuchung in Peru besonders interessant.[1]
Seit 1985 gibt es in Peru eine Richtlinie für die Durchführung von klinischen Studien, das erste Forschungsprojekt bekam 1995 grünes Licht. Bis 2015 wurden insgesamt 1.797 Studienprotokolle eingereicht, von denen 121 nicht genehmigt wurden. Mit der Inspektion von Studienzentren wurde erst 2004 begonnen. Diese Kontrollen führten zu zahlreichen Beanstandungen. Aber die VersuchsteilnehmerInnen selbst standen bisher nur selten im Fokus der Kontrolleure. Eine aktuelle Studie, die MitarbeiterInnen des peruanischen Gesundheitsministeriums und WissenschaftlerInnen aus den USA gemeinsam durchführten, will diese Lücke schließen.
Anlass der Untersuchung: Bei der Inspektion eines Studienzentrums im Jahr 2011, in dem das Diabetesmedikament Pioglitazon getestet wurde, fanden die InspektorInnen den Vermerk, dass alle PatientInnen über das neu bekannt gewordene Risiko von Blasenkrebs informiert worden waren und zugestimmt hatten, weiter an der Studie teilzunehmen. Unterschriebene Formulare waren allerdings nicht auffindbar. Ein Patient wurde daraufhin befragt und antwortete: „Ich hatte keine Ahnung von dem Risiko.“
Qualitative Befragung
Bei der aktuellen Untersuchung standen die Betroffenen im Mittelpunkt. Es wurden nach dem Zufallsprinzip 13 PatientInnen aus drei Studien zu TB-Medikamenten interviewt. Zunächst wurde gefragt, wie der Krankheitsverlauf vor der Teilnahme war. KeineR hatte die Symptome zunächst mit TB in Verbindung gebracht und drei PatientInnen erhielten zunächst eine falsche Diagnose. Die Behandlungsverläufe waren häufig nicht optimal, ein Studienteilnehmer hatte sich mit Eisentabletten selbst behandelt, drei hatten sich von Apothekenpersonal behandeln lassen. Zwei hatten eine TB-Behandlung vorzeitig abgebrochen und in der Folge eine multiresistente TB entwickelt. Eine Patientin hatte an einer Studie mit Standardbehandlung teilgenommen und wurde zu ihrem Entsetzen ausgeschlossen als sich herausstellte, dass sie an MDR-TB erkrankt war. Einige PatientInnen mit offener TB hatten einfach weitergearbeitet, obwohl sie teils bei ihrer Arbeit mit sehr vielen Menschen in Kontakt kamen.
(Keine) Zustimmung
Ein zentrales Kriterium für jede Studie ist, dass die Versuchspersonen der Teilnahme nach vollständiger Aufklärung über das geplante Vorgehen und die Risiken zustimmen müssen. Doch auch hier gab es offensichtliche Mängel: Zehn StudienteilnehmerInnen äußerten sich zum Aufklärungsformular, die Hälfte hatte es ungelesen unterschrieben. Ein Teilnehmer sagte: „Ich war so traurig als sie mir sagten, was ich habe. Ich dachte: macht mit mir, was ihr wollt. […] Wo muss ich unterschreiben? Zu der Zeit wusste ich nicht, was ich tue […]. Der Arzt sagte zu mir, wenn sein Sohn in der gleichen Lage wäre, würde er ihm die Teilnahme empfehlen.“ Mit dem Verständnis war es ebenfalls nicht weit her: „Sie gaben mir einige Seiten zum Unterschreiben. [Ich nahm ein Kopie nach Hause] ich verstand kein Wort, als ich einige Sätze las. […] Die Namen der Medikamente standen drin und ihre Wirkungen, die Namen der Ärzte, sechs an der Zahl, da waren auch die Namen von denen beim Ministerium, der Präsident, irgend so etwas [...].
Warum teilnehmen?
Wichtigstes Argument für die Studienteilnahme war die Hoffnung auf eine schnellere Heilung, gefolgt von besserer Betreuung und dass die Behandlung nichts kostet. Auch Versprechungen werden zitiert: „Ich bekäme eine individuelle Behandlung, nicht so wie im Gesundheitszentrum, wo alle auf dem Haufen sind, jeder weiß, dass du TN hast, sie kennen dich […] das ist nicht angenehm […] hier ist es persönlicher.“ „Es erhöhe meine Chancen auf Heilung von 55% auf 99%.“
Studie?
Kaum eineR der PatientInnen hatte wirklich verstanden, dass er oder sie an einer Studie mit einem nicht zugelassenen Medikament teilnahm. Eine Kranke meinte, dass ihr das niemand gesagt hätte, aber tief in ihrem Herzen wusste sie, „[…] dass sie mit meinem Körper experimentieren, damit sie mir helfen können, damit ich mich besser fühle.“ Der Begriff „Doppelblind“ wurde interpretiert als „nicht mehr sehen können und dass es etwas mit den Augen macht.“ Die meisten konnten mit dem Begriff Placebo nichts anfangen.
Dass es unerwünschte Wirkungen geben könnte, war kaum jemandem klar. Drei Personen informierten sich im Internet, und stellten fest, dass die Versuchsleiter etwas anderes gesagt hatten: „Ich fragte den Arzt, wie ist es mit den Nebenwirkungen? Ich weiß nicht, ob er meine Intelligenz oder die von allen beleidigte, [als] er sagte, keine […]. Sie werden keinerlei Probleme haben, im Gegenteil, sie werden schneller geheilt. […] Das ist nicht wahr; so ein starkes Medikament muss Folgen haben. […] Später als ich nachforschte [es folgt eine Liste von Nebenwirkungen].“
Schlecht informiert
Die mangelnde Aufklärung hatte aber auch potenzielle Folgen für die Zuverlässigkeit der Studienergebnisse. Den meisten war klar, dass sie die Behandlung jederzeit abbrechen, aber ansonsten den ärztlichen Empfehlungen folgen mussten, an den Untersuchungen teilnehmen und alle Probleme durch die Behandlung melden sollten, aber vier PatientInnen wussten nicht, an wen sie sich hätten wenden können. Den meisten war nicht bewusst, dass für sie eine Versicherung abgeschlossen war.
Nur ein Patient erwähnte, dass er keine anderen Medikamente einnehmen durfte. Einige änderten eigenmächtig ihre Medikation, teilten das aber den BehandlerInnen nicht mit. „Manchmal änderte ich meine Medikamente, weil ich sah, dass die Ärzte bei anderen Patienten, denen es besser ging oder die es nicht gut vertrugen, die Dosis von Amikacin reduzierten, ich machte das auch, weil ich mich schlecht fühlte […] und natürlich habe ich es ihm nicht gesagt […] das passierte drei Mal und ich hatte Recht.“
Der Mehrheit der Versuchspersonen war nicht richtig klar, ob sie an einer Studie zur besseren Versorgung von TB-PatientInnen teilnahmen oder an einem Medikamententest. Entsprechend war ihnen auch die Wichtigkeit, sich an die Medikationsvorschriften zu halten und Abweichungen mitzuteilen, weniger bewusst. So gesehen war das Versprechen einer fürsorglicheren Behandlung kontraproduktiv.
Die AutorInnen des Berichts stellen fest, dass den InspektorInnen viele relevante Dinge mitgeteilt wurden, die das Personal der Studie vorher nicht erfahren hatte. Die Kosten für solche Interviews seien gering und würden – wenn sie schon während der Durchführung der Studie gemacht werden – die Qualität und Zuverlässigkeit der Ergebnisse deutlich erhöhen. (JS)
Foto: US Navy 110503-N-QD416-125 Peruvian patients wait for eye care at a Continuing Promise 2011 medical clinic
Artikel aus dem Pharma-Brief 4-5/2018, S. 1
[1] Minaya GL et al. (2017) A Missing Piece in Clinical Trial Inspections in Latin America: Interviews With Research Subjects in Peru. Journal of Empirical Research on Human Research Ethics; 12, p 232
Resistente Keime in deutschen Gewässern
Kläranlagen machtlos gegen bakterielle Erreger
Vor etwa einem Jahr deckte der Norddeutsche Rundfunk auf, dass Flüsse und Trinkwasser in Hyderabad hochgradig mit resistenten Keimen verseucht sind.[1] Die indische Metropole ist eine Hochburg der Pharmaindustrie und die Abwässer sind stark mit Antibiotika belastetet. Neue Recherchen zeigen: Auch deutsche Gewässer sind betroffen.
Im Auftrag des NDR waren Proben aus 12 Flüssen, Bächen und Badeseen in Niedersachsen untersucht worden. Überall fand sich eine hohe Konzentration resistenter Keime. Grund dafür sind Gülle und Gärreste aus Biogasanlagen, mit denen antibiotische Rückstände auf die Felder und ins Wasser gelangen, aber auch Krankenhäuser und Pflegeheime. Der Leiter des Fachbereichs Nosokomiale Infektionen, Surveillance von Antibiotikaresistenz und -verbrauch im Robert Koch Institut (RKI) hält die Befunde für alarmierend.[2] Auch das Umweltbundesamt ist besorgt und bemängelt, dass es keine systematische Probenentnahme gibt.[3] Deutsche Kläranlagen sind ebenso wie indische nicht dafür ausgerüstet, resistente Keime aus Abwässern herauszufiltern. Zwar hält das Bundesumweltministerium eine milliardenschwere Nachrüstung für sinnvoll, doch zuständig sind die Länder.[4] Seit 2016 fördert das Bundesamt für Bildung und Forschung das Projekt HyReKA. Der wissenschaftliche Verbund untersucht die Verbreitung resistenter Keime im Abwasser und entwickelt verbesserte Aufbereitungstechnologien. Solche Innovationen weltweit verfügbar zu machen wäre essentiell für die globale Gesundheit - Antibiotika-Einträge zu reduzieren ebenfalls. (SK, CJ)
Artikel aus dem Pharma-Brief 2/2018, S. 2
[1] Schaaber J (2017) Resistente Keime in Indien. Pharma-Brief 5/2017, S. 1
[2] Baars C. Lambrecht O (2018) Gefährliche Keime in Bächen, Flüssen und Seen. NDR Panorama, 6.2.2018. www.ndr.de/nachrichten/niedersachsen/Gefaehrliche-Keime-in-Baechen-Fluessen-und-Seen,keime302.html
[3] Die Zeit (2018) Antibiotika-resistente Keime in Gewässern gefunden. 6 .2. 2018. www.zeit.de/news/2018-02/06/antibiotika-resistente-keime-in-gewaessern-gefunden-180206-99-948188 [Zugriff 20.2.18]
[4] NDR (2018) Fragen und Antworten zu Keimfunden in Gewässern. 6. 2. 2018. www.ndr.de/nachrichten/niedersachsen/Fragen-und-Antworten-zu-Keim-Funden-in-Gewaessern,keime304.html [Zugriff 20.2.18]
Nur die halbe Wahrheit
Empfehlungen zu Tropenkrankheiten mit Lücken
Was kann Deutschland zum Kampf gegen vernachlässigte Krankheiten beitragen? Eine Studie im Auftrag des Deutschen Netzwerks gegen vernachlässigte Tropenkrankheiten (DNTDs) gibt umfangreiche Empfehlungen.[1] Die meisten sind begrüßenswert, aber Interessenkonflikte, die die Umsetzung der Ziele konterkarieren könnten, werden ausgeblendet.
Das DNTDs ist ein Netzwerk verschiedener Forschungseinrichtungen, Nichtregierungsorganisationen und Pharmakonzerne, das auf eine Initiative des Verbandes forschender Arzneimittelunternehmen (Vfa) zurückgeht.[2] Ilona Kickbusch, eine bekennende Befürworterin von Public Private Partnerships,[3] ist Hauptautorin des Berichts.
In der internationalen gesundheitspolitischen Debatte spielt die Soziologin und Politikwissenschaftlerin Kickbusch eine zentrale Rolle. Im August 2017 berief Bundesgesundheitsminister Gröhe sie zur Vorsitzenden eines recht einseitig zusammengesetzten Fachgremiums, das sein Ministerium zu Fragen der globalen Gesundheit beraten soll (der Pharma-Brief berichtete).[3] Kickbuschs Standpunkte zu vernachlässigten Krankheiten kann man nun in der DNTDs-Publikation lesen.
Nachhaltige Entwicklungsziele
Zentrales Element der Empfehlungen ist es, die nachhaltigen Entwicklungsziele (Sustainable Development Goals SDGs) zu verwirklichen. Ein wichtiges Werkzeug dafür ist eine grundlegende und erschwingliche Gesundheitsversorgung für alle (Universal Health Coverage). Strategien zur Bekämpfung vernachlässigter Krankheiten dürften nicht länger bei einzelnen krankheitsbezogenen Programmen verharren, sondern müssten in umfassendere Ansätze integriert sein, so der Bericht. Vernachlässigte Krankheiten sollten als Querschnittsthema verstanden werden, das in allen Bereichen Beachtung findet – von der Armuts- und Hungerbekämpfung sowie der Trinkwasser- und Sanitärversorgung bis hin zur gleichberechtigten Teilhabe.
Dieser Ansatz ist an sich nicht neu und wird von vielen Akteuren gefordert. Auch die Pharma-Kampagne weist seit Jahrzehnten auf die Notwendigkeit integrierter Programme hin. Kickbusch sieht in der derzeitigen globalpolitischen Situation eine Chance zur Umsetzung der SDGs, aber bestehende Interessenkonflikte ignoriert sie.
Blick zurück
Das beginnt bei ihrer Bestandsaufnahme zur Bekämpfung vernachlässigter Krankheiten. Die teils unrühmliche Rolle der Industrie, was die Verfügbarkeit lebensrettender Therapien angeht, wird im historischen Rückblick ausgeblendet. Beispielsweise wurde 1995 das wichtige Medikament gegen Schlafkrankheit, Eflornithin, als „unrentabel“ vom Markt genommen, fünf Jahre später aber als lukratives Kosmetikum wieder eingeführt.
Systematische und koordinierte Strategien gab es erstmals um das Jahr 1952, als WHO und UNICEF ein Programm zur Frambösie starteten, einer bakteriellen Infektion. Bis in die 2000er Jahre war die globale Arbeit vor allem durch spezifische Krankheitsprogramme geprägt. Beispielhaft sind die präventiven medikamentösen Massenbehandlungen gegen Bilharziose, Wurmerkrankungen oder Flussblindheit. Weitere Säulen der WHO-Arbeit sind gezielte Behandlungsprogramme (z.B. bei Chagas, Schlafkrankheit, Lepra), die Vektorkontrolle zur Eindämmung von Krankheitsüberträgern (z.B. die Bekämpfung der Tigermücke, die u.a. das Dengue-Fieber überträgt) sowie veterinärmedizinische Maßnahmen, um die häusliche Viehhaltung zu verbessern (z.B. Echinokokkose). Das Programm WASH soll wiederum die Sanitär- und Trinkwasserhygiene verbessern und damit u.a. Wurmerkrankungen vorbeugen.
Einen Meilenstein verortet die Studie im Jahr 2006, da habe die Bündelung der Akteure begonnen. So wurden im USAID NTD-Programm erstmals Strategien gegen fünf vernachlässigte Krankheiten in einem Programm vereint. Die dritte große Veränderung sei 2015 mit den SDGs und dem Konzept der integrierten Programme gefolgt.
Auch die gesundheitspolitischen Rahmenbedingungen hätten sich zum Positiven verändert, so die AutorInnen. Neben dem erklärten Ziel der Universal Health Coverage werde auch globale Gesundheit immer mehr als wichtiges Thema wahrgenommen, wie die Beispiele Ebola und Zika zeigten. Und wirtschaftliche Veränderungen verschöben geopolitische Gewichte. China trete als neuer Akteur in Afrika auf, andere Schwellenländer wie Südafrika oder Brasilien würden immer stärker in die Verantwortung genommen.
Bestandsaufnahme
Als relevante Akteure machen die AutorInnen vor allem drei Gruppen aus: die Geldgeber (USA, Großbritannien, Gates), die Industrie (Arzneimittelspenden) und die Partnerländer, die das Thema vernachlässigte Krankheiten möglichst in ihre allgemeine Gesundheitsversorgung integrieren („ownership“) sollten. Dass der Einfluss einzelner Staaten und privater Geldgeber nicht unproblematisch ist, wird nicht weiter thematisiert (siehe Artikel auf S. 1).
Bei den bisherigen Programmen sehe die Bilanz sehr uneinheitlich aus. Bei der Schlafkrankheit (HAT) sei man auf dem Weg zur Eliminierung, wogegen Leishmaniose in Krisengebieten immer wieder aufflamme. Bei Chagas und anderen Krankheiten versagten die bisherigen Bekämpfungsstrategien sogar.
Von Seiten der staatlichen Entwicklungszusammenarbeit laufen derzeit zwei spezifische Programme. Die „Bekämpfung vernachlässigter Tropenkrankheiten in der CEMAC-Region“ fokussiert auf Zentralafrika, „Fit for School“ auf die Länder Indonesien, Kambodscha, Laos und Philippinen. Nicht-staatliche Programme organisieren unter anderem DAHW, DIFÄM, Christoffel Blindenmission und MSF.
Zur Forschungsförderung gibt es seit 2010 verschiedene Initiativen des Bundesministeriums für Bildung und Forschung, wobei die Finanzierung im internationalen Vergleich noch weiter ausgebaut werden sollte.
Wie weiter?
Der Bericht empfiehlt, Programme zu vernachlässigten Krankheiten stärker mit anderen Programmen (Landwirtschaft, Wasser etc.) zu verknüpfen. Als positives Beispiel wird die „BMZ Wasserstrategie“ (2017) genannt. Deren Ziel ‚Zugang zu Sanitär- und Trinkwasserversorgung schaffen und Hygiene sicherstellen‘ hat direkten Bezug zu vernachlässigten Krankheiten. Dort solle jedoch die Vektorkontrolle stärker berücksichtigt werden. Denn die Erfahrung zeige, dass es in der Folge großer Damm- und Wasserkraftwerkprojekte häufig zu einem (Wieder-)Aufflammen von Bilharziose (Schistosomiasis) komme (z.B. Gezira-Managil Dam, Sudan).
Auch ein Projekt aus Tansania wird positiv hervorgehoben Die Deutsche Lepra- und Tuberkulosehilfe und das Missionsärztliche Institut haben dort vernachlässigte Krankheiten, WASH-Komponenten (Trinkwasser, befestigte Bootsanleger), Aufklärungskampagnen und medikamentöse Behandlung von Bilharziose (Schistosomiasis) zusammengebracht.
Analoge Verknüpfungen wären auch für andere SDGs wichtig, etwa die Hunger-Bekämpfung oder Geschlechtergerechtigkeit. Deutschland müsse solche integrativen, vernetzten Ansätze nicht nur in den eigenen Programmen der Entwicklungszusammenarbeit verfolgen, sondern auch entsprechende multilaterale Aktivitäten z.B. bei der WHO unterstützen.
In Deutschland solle außerdem die Translationsforschung bei Diagnostika und Therapien ausgebaut werden, also die Weiterführung von Grundlagenforschung hin zur Produktentwicklung. Das läuft im Wesentlichen über die Förderung von Produktentwicklungspartnerschaften PDPs, aber auch beim Deutschen Zentrum für Infektionsforschung DZIF, einem deutschlandweiten Forschungsverbund.
Nicht genug
In einem zentralen Punkt liegt der Bericht richtig: Universal Health Coverage und Aufbau von Gesundheitssystemen sind wichtig. Damit geht er einen Schritt weiter als die „Londoner Erklärung“ von 2012, wo Pharmaunternehmen und Gates Stiftung ihre Sicht auf die Dinge vorstellten. Auf diese Erklärung beruft sich das DNTDs.[4] Die Stärkung lokaler Gesundheitssysteme war dort noch kein Thema.[5]
Schwächen zeigen sich, wenn die Rede auf multinationale Pharmaunternehmen kommt. Sie werden ausführlich für ihre Spenden gelobt, und es wird eine „NTD community“ beschrieben, die ein „sektorübergreifendes ‚Öko-system‘ aus NGOs, Unternehmen und Wissenschaft“ sei, das noch enger zusammenarbeiten solle. Den Pharmaunternehmen wird dabei eine „Mittlerrolle zwischen den Politikfeldern“ zugesprochen.
Pharmaindustrie als Mittler und Teil der Lösung? Sie ist eher Teil des Problems. Eine umfassende bedarfsgerechte Gesundheitsversorgung endet nicht damit, dass Medikamente für vernachlässigte Krankheiten gespendet werden. Auch die Behandlung von Krebs oder Hepatitis muss bezahlbar sein – um nur zwei Beispiele zu nennen, wo Pharmaunternehmen mit ungerechtfertigt hohen Preisen selbst für reiche Länder die Behandlungskosten in schwindelnde Höhen treiben. Hohe Produktkosten, Forschungslücken, Lieferengpässe, Intransparenz bei Studiendaten – die Liste der Probleme im regulären Pharmamarkt ist auch abseits des Themas vernachlässigte Krankheiten lang.[6]
Wenn wirtschaftliche Gewinninteressen mit den Bedürfnissen der Gesundheit kollidieren, muss das klar benannt werden. Denn einer Gesundheitsversorgung für alle, die schon 1978 die Erklärung von Alma Ata forderte, stehen bis heute zahlreiche mächtige Akteure und Partikularinteressen im Weg. Gerade eine „integrierte Umsetzung“ globaler Gesundheitsziele erfordert es, Gesundheitssysteme und -politiken umfassend in den Blick zu nehmen. Dazu gehört auch die kritische Betrachtung von Pharmaunternehmen als zentrale Akteure. Die enge Fokussierung auf einige Projekte zur Erforschung tropischer Krankheiten ist hier wenig zielführend. Aber da das Deutsche Netzwerk gegen vernachlässigte Tropenkrankheiten im Wesentlichen von der Pharmaindustrie gegründet wurde, sparen die AutorInnen andere Aktivitäten der Unternehmen wohlweislich aus. (CW)
Artikel aus dem Pharma-Brief 1/2018, S. 6
[1] Kickbusch I und Franz C (2017) Die integrierte Umsetzung der Bekämpfung der vernachlässigten Tropenkrankheiten – Potential Deutschlands. www.dntds.de/de/aktivitaeten-details/deutschlands-potential-bei-der-bekaempfung-von-vernachlaessigten-tropenkrankheiten.html
[2] Pharma-Brief (2013) Pharmaindustrie erfindet Zivilgesellschaft neu. Nr. 10, S. 6
[3] Pharma-Brief (2017) Deutschland: Einseitiger Rat: Nr. 7, S. 8
[4] www.dntds.de/de/hintergrund.html
[5] Pharma-Brief (2012) WHO oder Industrie? Nr. 1, S. 1
[6] Pharma-Brief (2016) 10 Mythen der Pharmaindustrie. Spezial Nr. 2
Memento-Preis
Engagement gegen TB ausgezeichnet
Der Memento Forschungspreis für vernachlässigte Krankheiten, den die BUKO Pharma-Kampagne gemeinsam mit Brot für die Welt, Ärzte ohne Grenzen und der Deutschen Lepra- und Tuberkulosehilfe verleiht, ging in diesem Jahr an Prof. Dr. Martina Sester von der Universität des Saarlandes und Prof. Dr. Dr. Christoph Lange vom Forschungszentrum Borstel.[1]
Forschungspreis
Beide engagieren sich im Forschungsnetzwerk TBnet für die Bekämpfung von Tuberkulose (TB) und eine bessere Versorgung von TB-PatientInnen europaweit. „Mit dem von Prof. Martina Sester und Prof. Christoph Lange aufgebauten Forschungsnetzwerk TBnet ist ein Zusammenschluss von Experten und Institutionen entstanden, die sich der Bekämpfung der TB in beispielhafter Weise widmen. (…) Den Ärztinnen und Ärzten vor Ort eine Ausbildung zu ermöglichen, eine verbesserte Diagnostik anzubieten und die notwendigen Medikamente zur Verfügung zu stellen, ist die große Leistung des TBnet und ein wichtiger Schritt, die Krankheit in Zukunft zu beherrschen“, sagte Jurymitglied Prof. Dr. August Stich, Chefarzt der Tropenmedizinischen Abteilung der Missioklinik Würzburg.
Journalistenpreis
Den Memento Journalistenpreis erhielt der Wissenschaftsjournalist Dr. Jakob Simmank. Mithilfe des Recherchestipendiums möchte er einen Beitrag über die Mesoamerikanische Nephropathie realisieren, einer noch unerklärlichen Epidemie der chronischen Nierenerkrankung, die vor allem ärmere Menschen in Lateinamerika betrifft. (CJ)
Artikel aus dem Pharma-Brief 2/2018
[1] http://memento-preis.de/memento-forschungspreis/s
MedizinerInnen fordern Klimaschutz
MedizinerInnen weltweit fordern einen wirksamen Klimaschutz und thematisieren die dramatischen Folgen der zunehmenden Erderwärmung auf die Gesundheit.
Im Oktober 2017 hatte der Weltärztebund (World Medical Association, WMA) wichtige Zielvorgaben gemacht: Auf seiner Generalversammlung in Chicago forderten die Mitglieder alle Regierungen dazu auf, die gravierenden gesundheitlichen Folgen des Klimawandels anzuerkennen und Aktionspläne zum Klimaschutz zu verabschieden. Nationale Ärztevereinigungen sollten sich für wirksame Regulierungsmaßnahmen wie eine CO2-Steuer oder Emissionshandelssysteme stark machen. Und auch bei der Entwicklung von politischen Konzepten, die die Auswirkungen des Klimawandels auf die Gesundheit minimieren, seien MedizinerInnen gefragt, so der WMA. ÄrztInnen seien darüber hinaus in der Pflicht, über den Klimawandel und seine globalen gesundheitlichen Folgen aufzuklären. Gemeinsam mit der WHO und anderen Akteuren sollten die nationalen Ärztevereinigungen geeignete Informationsmaterialien erstellen.[1]
Seither ist auch in Deutschland Bewegung in die Klima-Debatte gekommen: Organisationen und Beschäftigte aus dem Gesundheitssektor werden zunehmend aktiv und melden sich zu Wort. Die Kritik ist auch ganz oben angekommen. Jüngstes Beispiel ist ein Interview mit dem Präsidenten der Bundesärztekammer (BÄK), Frank Ulrich Montgomery im Umweltmagazin movum.[2] „Der Klimawandel ist real, er ist von Menschen gemacht und er gefährdet unsere Gesundheit.“ Montgomery betonte, dass es wichtig sei, das Gesundheitswesen in den Kampf gegen die Klimaerwärmung einzubinden. „Wir sprechen hier von rund 230.000 Einrichtungen – eine riesige Zahl, die wir über Multiplikatoren ansprechen wollen.“ ÄrztInnen sieht er aufgrund der dramatischen gesundheitlichen Folgen des Klimawandels ganz besonders in der Pflicht: „Deshalb sollte sich auch die Ärzteschaft für den Ausstieg aus den fossilen Brennstoffen engagieren.“
Zum Weltgesundheitstag am 7. April bezog nicht zuletzt die AG Klimawandel und Gesundheit des Netzwerks der Kritischen Mediziner*innen Stellung:[3] Ein schnellstmöglicher und sozialverträglicher Kohleausstieg sei unabdingbar. „Die katastrophalen gesundheitlichen Folgen der Kohleverstromung sind viel zu lange ignoriert worden“, schreibt das Netzwerk. Sein dezidiertes Positionspapier Gesundheit braucht Klimaschutz haben auch etliche Ärzte- und Umweltorganisationen sowie die Klima-Allianz Deutschland und die BUKO Pharma-Kampagne unterzeichnet.
Bereits Ende 2017 hatten sich Organisationen und Einzelpersonen aus dem Gesundheitsbereich in der Deutschen Allianz Klimawandel und Gesundheit zusammengeschlossen – auch die Pharma-Kampagne ist hier aktiv. Gemeinsames Ziel es ist, den Klimawandel als wichtiges Gesundheitsthema zu etablieren und politische und gesellschaftliche Veränderungen anzustoßen, um die weitere Erderwärmung auf deutlich unter 2°C zu begrenzen.[4] (CJ)
Artikel aus dem Pharma-Brief 3/2018, S. 6
[1] WMA (2018) Declaration of Delhi on Health and Climate Change. Adopted by the 60 WMA General Assembly, New Delhi, India, October 2009 and amended by the 68th WMA General Assembly, Chicago, United States, October 2017. www.wma.net/policies-post/wma-declaration-of-delhi-on-health-and-climate-change/ [Zugriff 17.4.2018]
[2] Müller M (2018) „ÄrztInnen sollen sich für den Fossil-Ausstieg engagieren“ Interview mit Ärztekammer-Präsident Frank Ulrich Montgomery. Movum Heft 19, 9/2018, S. 3 www.movum.info/images/ausgaben/heft19/heft19.pdf [Zugriff 17.4.2018]
[3] AG Klimawandel und Gesundheit der Kritischen Mediziner*innen Deutschland und und DNR (2018) Gemeinsame Pressemitteilung, 7.4.2018 www.vdaeae.de/index.php/themen/europeanhealthpolicy/929-gemeinsame-pressemitteilung-der-ag-klimawandel-und-gesundheit-der-kritischen-mediziner-innen-deutschland-und-dnr [Zugriff 17.4.2018]
[4] Deutsche Allianz Klimawandel und Gesundheit. www.klimawandel-gesundheit.de/ [Zugriff 17.4.2018]
Manifest zu den Europawahlen
Gesundheit hat Vorrang: Zugang zu Medikamenten in Europa verbessern
Viele Europäische Länder können ihrer Bevölkerung eine Gesundheitsversorgung auf hohem Niveau bieten, deren Kernprinzipien Gleichheit, Solidarität und Universalität sind.
Auch wenn es immer noch bedeutsame Ungleichheiten gibt, sowohl zwischen verschiedenen EU-Staaten als auch innerhalb der einzelnen Staaten, können wir dennoch grundsätzlich stolz auf diese Gesundheitsversorgung sein – besonders im Vergleich zum Viele ausgrenzenden und teuren US-Modell.
Gesundheitsversorgung ist ein öffentliches Gut, das von grundlegender Bedeutung für das Wohlergehen der Menschen ist. Es handelt sich um eines der wichtigsten Grundrechte, und die meisten EuropäerInnen wünschen, dass die EU mehr für die Gesundheit tut.[1] Zum Recht auf Gesundheit gehört der Zugang zu rechtzeitiger, akzeptabler und bezahlbarer Versorgung und zu qualitativ hochwertigen Medikamenten.[2] Mitgliedsstaaten sind verpflichtet, dieses Recht auf nicht diskriminierende Weise umzusetzen. Aber in den letzten Jahren haben wir uns bei der Produktion und der Preisgestaltung neuer Medikamente zunehmend einer Logik gebeugt, die auf zweistellige Gewinne ausgerichtet ist.
Viele Gesundheitssysteme in der EU leiden unter den Folgen einer einseitigen Industriepolitik und Regeln für geistige Eigentumsrechte im pharmazeutischen Sektor. Beide zielen fast ausschließlich auf das Wachstum der europäischen Wirtschaft und auf Gewinnmaximierung, statt die Versorgung der Bevölkerung mit guten und bezahlbaren Medikamenten in den Mittelpunkt zu stellen. In Europa und weltweit steigen die Preise neuer Arzneimittel schnell. Das bedeutet für die öffentlichen Gesundheitssysteme enorme finanzielle Belastungen. In der Folge wird eine steigende Zahl von Behandlungen für lebensbedrohende Infektionen und Krankheiten wie Krebs oder Hepatitis C für viele PatientInnen und viele nationale Gesundheitssysteme unbezahlbar.[3]
Das ist das Resultat eines ineffektiven und teuren Systems für Forschung und Entwicklung (F&E), das neue Medikamente mit Monopolen belohnt. Der Patentschutz versagt dabei, gesunden Wettbewerb zu fördern und wirkliche Innovationen zu belohnen.[4] Das derzeitige System erlaubt es Firmen, exorbitante Preise zu verlangen, die die Budgets öffentlicher Gesundheitsversorgung strapazieren. Geld, das an anderer Stelle fehlt. So gefährden sie die Nachhaltigkeit öffentlicher Gesundheitssysteme in Europa.[5]
Es müssen dringend Maßnahmen ergriffen werden, damit Regierungen und BürgerInnen Zugang zu bezahlbaren unentbehrlichen innovativen Medikamenten haben. Ein wichtiger Schritt in diese Richtung sind gemeinschaftliche Forschungsprozesse und das Teilen von Wissen. Die EU hat bereits Schritte in diese Richtung unternommen, indem sie Open Science und Open Innovation fördert. Diese Ansätze müssen ausgeweitet und auch in der biomedizinischen F&E umgesetzt werden.
Die anstehenden EU Wahlen sind eine Chance, Menschen und ihre Bedürfnisse in den Mittelpunkt der Europäischen Politik zu stellen. Das kann dazu beitragen, das Vertrauen der BürgerInnen in das Projekt Europa zu festigen, indem es zeigt: Die EU kümmert sich um das, was den Menschen am meisten bedeutet.
Gute Regeln für gesundheitsrelevante Forschung und Entwicklung sind ein wesentlicher Baustein des Projekts Europa. Um hier die BürgerInnen in das Zentrum der politischen Entscheidungen zu stellen, müssen folgende Themen für das nächste Europäische Parlament und die Europäische Kommission höchste Priorität bekommen:
Public return on public investment: Das Geld der SteuerzahlerInnen, das in biomedizinische F&E investiert wird, sollte eine öffentliche Rendite bringen und der Gesellschaft nützen. EU Investitionen müssen sich an den Bedürfnissen der öffentlichen Gesundheit orientieren, und die Forschungsergebnisse müssen zugänglich, verfügbar und bezahlbar sein. Open Science, Open Data und Zugang zu den wissenschaftlichen Veröffentlichungen sollten zum Standard werden.
Ein nachhaltiges System der Forschung und Entwicklung: Die EU und ihre Mitgliedsstaaten sollten gesunden Wettbewerb fördern und wirkliche Innovation belohnen. Das schafft ein nachhaltiges System für Regierungen und PatientInnen. Neue Modelle der F&E, die auf Open Science Prinzipien basieren, sollten erprobt werden. Beispiele sind De-Linkage-Modelle, die Arzneimittelentwicklung von der Aussicht auf hohe Preise abkoppeln, Forschungsprämien, sozialverträgliche Lizenzierungen und Open Source Forschung. Solche Modelle sollten mit Pilotprojekten, Machbarkeitsstudien und neuen Finanzierungsprogrammen gefördert werden.
Gesunder Wettbewerb und Handel: Die EU sollte ihr System für geistige Eigentumsrechte reformieren, um eine gesunde Balance zwischen privaten und öffentlichen Interessen zu erreichen. Wettbewerb ist wichtig, um echten Fortschritt zu fördern: Die EU sollte einen fairen Wettbewerb ermöglichen, indem sie wettbewerbswidrigen Praktiken entgegenwirkt und sie auch sanktioniert. Zudem sollte es die EU unterlassen, unfaire Standards für geistige Eigentumsrechte zu exportieren, Stattdessen sollte sie die Handelspolitik so ausrichten, dass sie der öffentlichen Gesundheit weltweit dient.[6]
Wirkliche Innovation und Sicherheit der PatientInnen: Neue innovative Arzneimittel müssen einen therapeutischen Zusatznutzen im Vergleich zu existierenden Behandlungsmöglichkeiten bieten. Hohe Standards für die wissenschaftliche Prüfung der Marktzulassung müssen gesichert und gefördert werden. Die Transparenz klinischer Studiendaten und die Risikoüberwachung von Arzneimitteln müssen verbessert werden. Europaweite Zusammenarbeit bei der Nutzenbewertung von Gesundheitstechnologien (Health Technology Assessment) sollte ausgebaut werden, wobei die Entscheidungen auf Evidenz beruhen müssen und auf einer größtmöglichen Transparenz und Unabhängigkeit basieren sollten. Jede Art von Interessenkonflikt sollte ausgeschlossen werden.
Dieses Manifest wurde in der European Alliance for Responsible R&D and Affordable Medicines entwickelt.
Original: http://medicinesalliance.eu/wp-content/uploads/2018/11/EP_Manifesto_English.pdf Übersetzung: BUKO Pharma-Kampagne
Unterstützende Organisationen:
- TranspariMED
- Wemos Foundation
- Commons Network
- EKPIZO
- Salud por Derecho
- Global Health Advocates – GHA
- Health Action International – HAI
- Acceso Justo al Medicamento, AAJM
- T1 International
- AIDES
- PRAKSIS
- Health and Trade Network
- Aidsfonds
- NoGracias (Spain)
- GAT - Grupo de Ativistas em Tratamentos
- ARAS - the Romanian Association Against AIDS
- International Society of Drug Bulletins (ISDB)
- Health Projects for Latvia
- Policies for Equitable Access to Health
- Verein demokratischer Pharmazeutinnen und Pharmazeuten (VdPP)
- Prescrire
- Médecins du Monde
- BUKO Pharma-Kampagne
- European Network against Privatization and Commercialization of Health and Social Protection
- Platform for Action on Health and Solidarity (Belgium)
Artikel aus dem Pharma-Brief 10/2018, S. 6
[1] 70% der EuropäerInnen wollen, dass die EU mehr für Gesundheit macht – so eine Umfrage von Eurobarometer im März 2017 www.europarl.europa.eu/external/html/eurobarometer-052017/
[2] Gesundheitsversorgung ist auch einer der 20 Grundsätze der europäischen Säule sozialer Rechte https://ec.europa.eu/commission/priorities/deeper-and-fairer-economic-and-monetary-union/european-pillar-social-rights/european-pillar-social-rights-20-principles_de#kapitel-iii-sozialschutz-und-soziale-inklusion
[3] Nach einer Schätzung von 2016 würde die Behandlung von 55% der Menschen mit chronischer Hepatitis C in Frankreich mehr kosten als das Budget aller öffentlichen Krankenhäuser in Paris. www.unsgaccessmeds.org/inbox/2016/3/4/pauline-londeix-enligsh-translation
[4] RVS Development of new medicines. Better, faster, cheaper. 2017 www.raadrvs.nl/uploads/docs/Recommendation_Development_of_New_Medicines.pdf
[5] Collier R. Drug development cost estimates hard to swallow. Canadian Medical Association Journal 2009;180(3): 279. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2630351
Prasad V, Mailankody S. Research and Development Spending to Bring a Single Cancer Drug to Market and Revenues After Approval. JAMA Intern Med. 2017;177(11):1569–1575. doi:10.1001/jamainternmed.2017.3601 https://jamanetwork.com/journals/jamainternalmedicine/article-abstract/2653012
Médecins Sans Frontières. Lives on the Edge: Time to Align Medical Research and Development with People’s Health Needs. May 2016. Page 13. Available from: www.msfaccess.org/sites/default/files/R&D_report_LivesOnTheEdge_Updated29Sept_ENG_2016.pdf
[6] Die Nutzung von TRIPS Flexibilitäten sollte in Europa ausgeweitet und außerhalb der EU gefördert werden.
Kritiker rausgeworfen
Warum Peter Gøtzsche aus Cochrane ausgeschlossen wurde
Der dänische Mediziner Peter Gøtzsche war bei Cochrane von Anfang an dabei. Im September wurde er mit knapper Mehrheit ( 6 : 5, 1 Enthaltung) aus dem Vorstand ausgeschlossen, kurz darauf wurde ihm die Mitgliedschaft aberkannt. Dahinter steckt mehr als ein angeblich zu rüder Ton.
Die Cochrane Collaboration wertet die Ergebnisse klinischer Studien in systematischen Übersichtsarbeiten (Systematic Reviews) aus und spielt deshalb eine wichtige Rolle in der Bewertung des Nutzens von Arzneimitteln. Dabei war die Organisation nicht immer frei von Interessenkonflikten. Bis vor 15 Jahren konnten sogar Hersteller Reviews finanzieren, was zu zweifelhaften Ergebnissen führte.[1] Aber auch die aktuellen Statuten der Organisation von 2014 schließen Probleme nicht aus, Personen mit Interessenkonflikten dürfen im Review-Team lediglich nicht die Mehrheit stellen.
Peter Gøtzsche leitete das nordische Cochrane-Zentrum in Kopenhagen. Erst 2017 war er in den Cochrane-Vorstand gewählt worden. Bei seiner Vorstellung hatte er die klare Ansage gemacht, dass er sich für striktere Regeln einsetzen werde. Das bescherte ihm die meisten Stimmen. Dass Gøtzsche Ernst machte, hat aber manchem in der Leitungsstruktur von Cochrane nicht gefallen. So kritisierte er im Sommer detailliert den Review zur HPV-Impfung wegen fehlender Daten und Interessenkonflikten der AutorInnen.[2]
Die Cochrane-Leitung machte den Rauswurf aber nicht an inhaltlicher Kritik fest, sondern an angeblichem persönlichen Fehlverhalten wie „schlechtem Benehmen“ oder der unberechtigten Nutzung des Namens Nordic Cochrane Center bei bestimmten Anlässen. Die Auseinandersetzung ist inzwischen in weiten Teilen öffentlich.[3] Obwohl die Cochrane-Leitung schwere persönliche Vorwürfe gegen Gøtzsche erhebt, weigert sie sich, Beweise für das Fehlverhalten vorzulegen – angeblich, um die Interessen Dritter, aber auch Gøtzsches zu schützen. Einblick in diese Argumentation bietet die Dokumentation eines Webinars, das die Cochrane-Leitung als Reaktion auf die breite Kritik an Gøtzsches Rauswurf veranstaltete.[4]
Am 28. September sagte sich Gøtzsche von Cochrane los und erklärte das Nordische Cochrane Center für unabhängig. Die Leitung von Cochrane wandte sich daraufhin an die dänische Regierung, die das Center finanziert.4 Anfang November wurde Gøtzsche ohne Begründung gekündigt und die Leitung des Nordischen Cochrane Center wurde ihm entzogen,[5] obwohl WissenschaftlerInnen aus aller Welt, darunter bekannte Personen wie John P.A. Ioannidis,[6] die dänische Gesundheitsministerin aufgefordert hatten, diese Entscheidung noch einmal zu überdenken.
Bleibt zu hoffen, dass der Vorfall wenigstens dazu führt, dass die Unabhängigkeit Cochranes intensiver debattiert wird. David Hammerstein, der aus Protest gegen Gøtzsches Rauswurf aus dem Cochrane-Vorstand zurücktrat, hat die Ereignisse in einem lesenswerten Beitrag zusammengefasst. Der Titel könnte Programm sein: „Erneuert Cochrane, um die Produktion von vertrauenswürdiger Evidenz zum Nutzen des Gemeinschaftsguts öffentlicher Gesundheit zu stärken.“[7]
Artikel aus dem Pharma-Brief 8-9/2018, S.7
Bild Peter Gøtzsche © Jörg Schaaber
[1] arznei-telegramm (2018) Was ist los im Cochrane-Leitungsgremium?; 49, S. 82
[2] Pharma-Brief (2018) PHV-Impfung: Verzerrte Übersicht. Nr. 7, S. 8
[3] Nachzulesen auf der Website von Peter Gøtzsche mit zahlreichen links zu Dokumenten: www.deadlymedicines.dk
[4] Cochrane (2018) Ohne Titel. https://community.cochrane.org/sites/default/files/uploads/Governing%20Board%20Webinars%20Oct%202018%20Questions%20and%20Answers.pdf
[5] Bro N (2018) Professorer laver underskriftsindsamling til støtte for Gøtzsche. Sundhedspolitisk Tidsskrift. 6. Nov. https://sundhedspolitisktidsskrift.dk/nyheder/1472-professorer-laver-underskriftsindsamling-til-stotte-for-gotzsche.html
[6] https://7905d1c4e12c54933a44d19fcd5f9356-gdprlock/sgdambrauskas/status/1064109964296372225/photo/1
[7] Hammerstein D (2018) Regenerate Cochrane to strengthen the production of trusted evidence for the common good of public health. No gracias, 8 Okt. www.nogracias.eu/2018/10/08/regenerate-cochrane-to-strengthen-the-production-of-trusted-evidence-for-the-common-good-of-public-health-by-david-hammerstein [Zugriff 19.11.2018]
Krebserregende Valsartan-Präparate
Warum die Kontrollen nicht ausreichen
Der Rückruf von verunreinigten Valsartan-Medikamenten ging im Sommer durch die Presse. In dem Blutdrucksenker war der krebserregende Stoff Nitrosamin entdeckt worden. Was steckt hinter diesem Skandal und was müsste passieren, um Wiederholungen unwahrscheinlicher zu machen?
Über 100 Valsartan-Chargen von 16 Herstellern waren allein in Deutschland von dem Rückruf betroffen.[1] Der in den Medikamenten enthaltene Wirkstoff stammte hauptsächlich von dem chinesischen Hersteller Zhejiang Huahai Pharmaceutical. Da die globale Produktion von aktiven Wirkstoffen hauptsächlich in China und Indien stattfindet, ist die Herkunft der verunreinigten Rohstoffe kein Zufall. Die Hersteller hierzulande machen meist nur die letzten Verarbeitungsschritte, tragen aber natürlich die rechtliche Verantwortung für die Qualität ihrer Produkte.
Risiko Krebs
Die Verunreinigung mit krebserregenden Nitrosaminen war erheblich: Die betroffenen Medikamente enthielten zwischen 4 und 22 µg NMDA (Nitrosodimethylamin) pro Tablette.[2] Zum Vergleich: Die durchschnittliche Aufnahme von Nitrosaminen durch Lebensmittel wird auf 0,3 µg pro Tag geschätzt.[3] Es gibt keinen Grenzwert, unterhalb dessen Nitrosamine als unbedenklich gelten.
Möglicherweise sind die Präparate schon seit 2012 verunreinigt. Viele PatientInnen hätten dann jahrelang krebserzeugende Medikamente geschluckt. Die europäische Arzneimittelbehörde EMA schätzt das Krebsrisiko auf 1 von 5.000 PatientInnen. Das heißt, von 5.000 Betroffenen, die das verunreinigte Präparat sieben Jahre in der höchsten Tagesdosis (320 mg) eingenommen haben, wird eine/r an Krebs erkranken.[4] Dabei ging die EMA allerdings von einer Belastung von 60 µg NMDA aus, in 320 mg Tabletten wurden in Deutschland maximal 22 µg gefunden.
Die US-Behörde FDA errechnete aus den tatsächlich in den belasteten Tabletten gefundenen Mengen bei vierjähriger Einnahme der höchsten Tagesdosis einen zusätzlichen Krebsfall auf 8.000 PatientInnen.[5] Diese beiden Berechnungen sind nicht stimmig und zeigen, dass seriöse Abschätzungen schwierig sind.
Das zusätzliche Krebsrisiko scheint zwar eher gering, auch wenn jeder unnötige Fall einer zu viel ist. Aber da allein in Deutschland über zwei Millionen Menschen Valsartan-Präparate einnehmen [6],[7] und die Verunreinigung vermeidbar ist, ist die späte Entdeckung der schädlichen Stoffe und die zögerliche Reaktion der Behörden inakzeptabel.
Mängel auf vielen Ebenen
Die Verunreinigung von Valsartan ist wohl mehr zufällig bei Kontrollen für die Produktion eines spanischen Fertigarzneimittelherstellers aufgefallen.[8] Die Überwachungsbehörde eines Bundeslandes erfuhr das durch einen anonymen Hinweis 3 und informierte die deutsche Zulassungsbehörde BfArM, die wiederum die europäische EMA alarmierte. Und hier fängt das Wirrwarr an. Obwohl die meisten Wirkstoffe aus ausländischen Quellen stammen, sind die Bundesländer für die Überwachung der Hersteller zuständig. Sie sind auch diejenigen, die eine Rückholung von Arzneimitteln anordnen können. Das BfArM kann nur eine koordinierende Rolle spielen.
Deshalb dauerten Testung und Rückrufe auch vergleichsweise lange. Kritik gab es auch an der Informationspolitik, die unvollständig und zögerlich war. So erfuhren ÄrztInnen und PatientInnen nur nach und nach, welche Produkte betroffen waren. Und der Hinweis, das Medikament nicht abzusetzen, sondern auf einen anderen Hersteller auszuweichen, lief ins Leere, weil keine Liste der unbedenklichen – weil negativ getesteten – Mittel veröffentlicht wurde.[1]
Hersteller mitverantwortlich
Auch die Hersteller in Deutschland trifft ein gehöriges Maß an Mitverantwortung, denn sie sind für die Qualität des Endprodukts verantwortlich, auch wenn sie den eigentlichen Wirkstoff selbst einkaufen. Sie erhalten vom Wirkstoffproduzenten Anweisungen zur Analyse und Qualitätskontrolle (Active substance master file, ASMF). Allerdings gibt es einen vertraulichen Teil, der den Syntheseweg offenlegt. Dieser steht nur den Zulassungsbehörden zur Verfügung.[3] Warum ist das ein Problem?
Die Verunreinigung von Valsartan bei Zhejiang Huahai Pharmaceutical ist durch eine Veränderung des Herstellungsprozesses entstanden, der eine höhere Wirkstoffausbeute bringen sollte. Doch genau diese Veränderung ließ bedeutende Mengen von Nitrosaminen entstehen. Ein erfahrener Chemiker hätte das Risiko durch den geänderten Syntheseprozess also erkennen und entsprechende Untersuchungen veranlassen können, bevor die Tabletten gepresst und an die Apotheken ausgeliefert wurden. Doch die Geheimhaltung des Herstellungsprozesses verhindert, dass die Fertigarzneimittelhersteller solche Risiken vollständig erkennen können.
Europäische Behörde versagte
Auf dem Weg von der Produktionsstätte eines Wirkstoffs bis zu seiner Weiterverarbeitung in Europa gibt es noch eine wichtige weitere Kontrollinstanz: Das beim Europarat angesiedelte EDQM[9] ist für die Sicherheit von pharmazeutischen Rohstoffen zuständig. Jeder Wirkstoff-Produzent benötigt für den Import nach Europa eine Erlaubnis für den Herstellungsprozess.[10] Aber offensichtlich hat die Behörde geschlafen. Denn Zhejiang Huahai Pharmaceutical hatte (wenn auch vermutlich verspätet) den geänderten Syntheseprozess für Valsartan offiziell gemeldet. Das Herstellungsverfahren wurde am 9.6.2016 vom EDQM genehmigt. Die Behörde erkannte die Risiken nicht.
Doch damit nicht genug: Die EDQM brüstete sich noch im August dieses Jahres, dass die Zusammenarbeit der nationalen Labore zur Qualitätskontrolle in Europa sehr gut funktioniere. Es seien 2017 rund 9.000 Arzneimittel getestet worden. Schwerpunkte der Kontrolle waren zehn Wirkstoffe, darunter auch Valsartan.[11] Dummerweise wurde die Verunreinigung mit Nitrosaminen trotzdem nicht bemerkt.
Mehr Kontrolle nötig
Inzwischen entzog das EDQM drei weiteren Herstellern die Erlaubnis für die Einführung von Valsartan nach Europa, zwei davon stammen aus China, einer aus Indien. Die geprüften Wirkstoffe enthielten entweder NDMA (wenn auch deutlich weniger als der von Zhejiang Huahai Pharmaceutical) oder es gab Zweifel an der Eignung des Herstellungsprozesses.[12]
Außerdem teilte das EDQM mit, dass die Untersuchungsbehörden in den europäischen Staaten jetzt parallel zwei unterschiedliche Testverfahren zur Bestimmung von Nitrosaminen in Valsartan anwenden und auch andere Wirkstoffe der Sartangruppe untersucht werden.
Möglicherweise ist der Sartan-Skandal nur die Spitze des Eisbergs. Eine bessere Kontrolle der Arzneimittelqualität ist dringend erforderlich. Eine Bündelung der Aufsicht beim BfArM ist erforderlich. Bei Gefahr im Verzug sollte die deutsche Behörde bevollmächtigt sein, Arzneimittel mit Qualitätsmängeln sofort vom Markt zu nehmen. Das EDQM muss die Qualität der eigenen Arbeit überprüfen und die Kontrolldichte erhöhen. In den ersten sieben Monaten dieses Jahres führte die Behörde ganze 22 Vor-Ort-Inspektionen in Asien durch.[13] Und schließlich müssen auch die Firmen, die die Wirkstoffe weiterverarbeiten, über Änderungen im Herstellungsprozess informiert werden, damit sie gezielt nach Verunreinigungen suchen können. (JS)
Artikel aus dem Pharma-Brief 7/2018, S.4
[1] arznei-telegramm (2018) Mehr als 100 Valsartan-Präparate mit Kanzerogen kontaminiert. 49, S. 65
[2] 160 mg Tabletten 4-10 µg; 320 mg Tabletten 16-22 µg
[3] Arzneimittelbrief (2018) Produktionsbedingte Kontamination von Valsartan-Präparaten: Weitere Informationen. 52, S. 57
[4] EMA (2018) Update on review of recalled valsartan medicines. Press release 2 Aug. www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2018/08/news_detail_003000.jsp
[5] FDA (2018) FDA update on valsartan recalls. www.fda.gov/Drugs/DrugSafety/ucm613916.htm [Zugriff 13.9.2018]
[6] Valsartan mono 717 Mio. Tagesdosen, in Kombinationen 241 Mio. Tagesdosen
[7] Schwabe U und Paffrath D (Hrsg.) (2017) Arzneiverordnungs-Report 2017. S. 227ff.
[8] BfArM (2018) Valsartan. Fragen und Antworten. www.bfarm.de/DE/Arzneimittel/Arzneimittelzulassung/Arzneimittelinformationen/Arzneimittelfaelschungen/RapidAlertSystem/Valsartan/Hintergruende/_node.html [Zugriff 13.9.2018]
[9] European Directorate for the Quality of Medicines & Healthcare (EDQM) www.edqm.eu
[10] Certificate of suitability (CEP)
[11] EDQM (2018) Factsheet OMCL Network. August www.edqm.eu/sites/default/files/report-cep-monthly-report-july2018.pdf
[12] EDQM (2018) Update on EDQM’s actions following detection of impurity in valsartan. 28 Aug. www.edqm.eu/sites/default/files/pressrelease-update-on-edqm-actions-following-detection-of-impurity-in-valsartan-august2018.pdf [Zugriff 13.9.2018]
[13] EDQM (2018) Certification monthly report July 2018. www.edqm.eu/sites/default/files/report-cep-monthly-report-july2018.pdf [Zugriff 13.9.2018]
Klimawandel kommt uns teuer zu stehen
Sonderbericht der WHO zur COP24
Lebensräume, Atemluft, Trinkwasser und Ernährung sind durch steigende Meeresspiegel, Extremwetter-Ereignisse, Hitzewellen und Dürren in Gefahr. Unterlassener Klimaschutz kommt uns teuer zu stehen. Mit ihrem Bericht zur Weltklimakonferenz COP24 in Kattowitz legt die WHO dazu aktuelle Berechnungen vor.[1]
Kostspieligen Maßnahmen zum Klimaschutz stehe ein gesundheitlicher Nutzen von doppeltem Wert gegenüber, resümiert die WHO in ihrem bei der Weltklimakonferenz vorgestellten Bericht. Auf 38 Seiten präsentiert ein 80-köpfiges internationales ExpertInnenteam seine Einschätzungen und Berechnungen.
Eine der Kernaussagen: In den 15 Ländern mit den höchsten Treibhausgas-Emissionen betragen die daraus resultierenden Gesundheitskosten mehr als 4 % ihres Bruttoinlandsproduktes. Maßnahmen zur Umsetzung des Pariser Abkommens kosten dagegen etwa ein Prozent des weltweiten BIP.[2] Klimaschutz mache sich daher überall auf der Welt bezahlt. Die zu erwartenden positiven Gesundheitseffekte von entschiedenem Handeln wären gerade in Indien und China besonders groß.
Der Bericht präsentiert den aktuellen Wissensstand zu den komplexen Zusammenhängen zwischen Klimawandel und Gesundheit und bietet Schlüsselinformationen für politische EntscheidungsträgerInnen: Wie ziehen Länder den größten gesundheitlichen Nutzen aus ihren Klimaschutzmaßnahmen und wie können die schlimmsten krankmachenden Folgen des Klimawandels vermieden werden? Der Bericht gibt außerdem einen Überblick über gesundheitspolitische Initiativen und Maßnahmen auf lokaler, nationaler und globaler Ebene zur Umsetzung der Paris-Konvention.
Jetzt Handeln!
Würde das Paris-Abkommen in Kattowitz konsequent umgesetzt, könnte es „die stärkste Gesundheits-Vereinbarung dieses Jahrhunderts sein“, sagte WHO-Direktor Dr. Tedros Adhanom Ghebreyesus. Der Klimawandel bedrohe schon heute die Grundlagen einer gesunden Existenz: Saubere Luft, Trinkwasser, Nahrung und eine sichere Unterkunft. Er drohe Jahrzehnte des Fortschrittes in der globalen Gesundheit zunichte zu machen. „Wir können es uns nicht leisten, das Handeln noch länger hinauszuzögern.“ Gerade für die Inselstaaten im Pazifik sei ein schnelles Handeln von essenzieller Bedeutung. Die Ergebnisse der COP24 entscheiden über Gesundheit und Existenz der InselbewohnerInnen.
Wetterextreme und Dürren rufen Hungersnöte hervor, Trinkwassermangel verursacht Krankheiten, führt zu mangelnder Hygiene und beeinträchtigt in erheblichem Maß die Mutter-Kind-Gesundheit. Hitzewellen lassen die Sterberaten bei Herz- und Atemwegs-Erkrankungen ansteigen und fördern Asthma-Anfälle. Denn die Konzentration von Schadstoffen, Pollen und Allergenen in der Luft ist bei Hitze deutlich erhöht.
Luftverschmutzung tötet
Die Folgen einer verfehlten Klimapolitik wären verheerend. Eine eher konservative Schätzung geht ab 2030 von jährlich 250.000 zusätzlichen Todesfällen durch den Klimawandel aus: 38.000 Sterbefälle weltweit durch Hitze, 48.000 durch Diarrhö, 60.000 durch Malaria und 95.000 durch Unterernährung bei Kindern.
Eine Einhaltung der Pariser Klimaschutzziele – die in vielen Bereichen eine Verbesserung der gegenwärtigen Situation bedeuten würde – könnte ab 2050 jedes Jahr eine Million Menschenleben retten – und zwar allein durch eine Reduzierung der Luftverschmutzung, die jedes Jahr für sieben Millionen vorzeitige Todesfälle sorgt. 90 % der globalen Stadtbevölkerung atmet derzeit Luft ein, deren Schadstoffbelastung die WHO als bedenklich einstuft.
Die Krankheitsbürde durch CO2-Emissionen sei inzwischen dermaßen hoch, dass ein Wechsel zu nachhaltigeren Energien, Transport- und Lebensmitteltechnologien sich ganz von allein auszahle. „Die wahren Kosten des Klimawandels sehen wir in unseren Krankenhäusern und fühlen wir in unseren Lungen“, formuliert es Dr. Maria Neira, Leiterin der WHO-Abteilung für umweltbedingte und soziale Determinanten von Gesundheit. Klimaschutz sei darum kein Kostenfaktor, sondern eine Chance.
Ein Plus für die Gesundheit
Investitionen in den Klimaschutz seien immer auch ein Plus für die Gesundheit. Denn dieselben Faktoren, die das Weltklima destabilisieren, sind auch verantwortlich für schlechte Gesundheit. Haupttreiber des Klimawandels ist die Verbrennung fossiler Energieträger – sie ist auch eine der Hauptursachen für Luftverschmutzung.
Fleischproduktion ist verantwortlich für 15% der CO2-Emissionen. Eine Reduktion der Fleischproduktion sowie des Fleischanteils in der täglichen Nahrung könnte das Risiko für Herz-Kreiskauf-Erkrankungen und Krebs deutlich senken.
Städte als Schlüsselakteure
Gerade Städte sieht die WHO in der Verantwortung, was den Klimaschutz angeht, sieht sie aber auch als besonders große Nutznießer effektiver Klimapolitik: [3] Denn die Weichen für viele der notwendigen Maßnahmen im Bereich Verkehr und Energieversorgung werden gerade auf lokaler Ebene gestellt. Zugleich profitiert gerade die städtische Bevölkerung überproportional von umweltfreundlichen Verkehrs- und Energiekonzepten. Eine Fußgänger- und Fahrrad-freundliche Verkehrspolitik fördert körperliche Aktivität und bedeutet eine bessere Gesundheit. Ein sicherer öffentlicher Nahverkehr senkt die Zahl der Verkehrsopfer und Unfälle.
Fehlende Investitionen lassen die Verletzlichsten zurück
Nicht nur beim Klimaschutz, auch bei der Klimaanpassung sieht die WHO gewaltige Lücken. Was zu tun ist, um die Gesundheit vor dem Klimawandel zu schützen, sei zwar bekannt – von krisenfesten, nachhaltigen Gesundheitseinrichtungen bis hin zu verbesserten Warnsystemen für Extremwetter-Ereignisse und Krankheitsepidemien. „Aber fehlende Investitionen lassen die Verletzlichsten zurück”, so Dr. Joy St John, aus der WHO-Abteilung für umweltbedingte und soziale Determinanten von Gesundheit.[2]
Der WHO-Bericht fordert alle Partnerstaaten der United Nations Framework Convention on Climate Change (UNFCCC) auf, eine Analyse von Gesundheitskosten und -nutzen in ihre Klimaschutz-Strategien einzubeziehen. Er empfiehlt fiskale Anreize wie eine Kohlesteuer oder Energie-Subventionen, um der Wirtschaft Anreize für eine Reduzierung von Treibhausgasen und Luftschadstoffen zu bieten. Nicht zuletzt sollten die Staaten in eine effektive Klimaanpassung im Gesundheitssystem investieren und ihre Infrastruktur auf zukünftige Herausforderungen besser vorbereiten.
Klimaschutz stärkt Entwicklung
Die Klimaschutz-Agenda sei nicht nur eng verzahnt mit der Agenda 2030 der nachhaltigen Entwicklungsziele, sondern letztendlich auch mit der internationalen Charta der Menschenrechte. Die WHO sieht alle Staaten in der Verantwortung, das Menschenrecht auf Gesundheit zu respektieren, zu schützen und zu erfüllen. Das erfordere eben auch, Menschen vor den vorhersehbaren und krankmachenden Auswirkungen des Klimawandels zu bewahren. (CJ)
Schulmaterial zum Klimawandel
Im Fokus dieser kostenlosen Unterrichtseinheit für Berufsschulen stehen Atemwegs-Erkrankungen in Indien und Deutschland. Die Broschüre mit Unterrichtskonzept und Arbeitsbättern bereitet die Schülerinnen und Schüler auf zukünftige berufliche Herausforderungen vor und sensibilisiert sie für die komplexen Gesundheitsrisiken durch den Klimawandel. Sie können das Material kostenlos herunterladen.
Artikel aus dem Pharma-Brief 10/2018, S. 1
Bild © Karolina Sobel Akcja Demokracja COP24
[1] WHO (2018) COP24 Special report: Health & Climate Change. https://apps.who.int/iris/bitstream/handle/10665/276405/9789241514972-eng.pdf?ua=1 [Zugriff 12.12.18]
[2] WHO (2018) Health benefits far outweigh the costs of meeting climate change goals. Press release 5. Dec www.who.int/news-room/detail/05-12-2018-health-benefits-far-outweigh-the-costs-of-meeting-climate-change-goals [Zugriff 12.12.18]
[3] WHO (2018) Health and Climate Change. Newsroom. Facts in pictures. www.who.int/news-room/facts-in-pictures/detail/health-and-climate-change [Zugriff 12.12.18]
Härtere Bandagen gegen Soft-Drinks
Großbritannien führt Zuckersteuer ein
Im Kampf gegen Übergewicht und Diabetes setzt nun auch die Regierung in London auf Härte. Mit einer Steuer auf Softdrinks greift sie eine wichtige Ursache an - die Hersteller reagieren. Auch in Deutschland wird die Debatte aufmerksam verfolgt, aber die Politik bleibt untätig.
„Britain is the fat man of Europe“ – so brutal wie die britische Times titelten viele Blätter im Königreich Ende 2017.[1] Neue OECD-Zahlen zeigten damals einen rasanten Anstieg von Übergewicht und Diabetes auf der Insel. Der Regierung wurde Tatenlosigkeit vorgeworfen, ein Aktionsplan zu Übergewicht bei Kindern war ein Jahr zuvor noch verwässert worden und zog deshalb harsche Kritik auf sich.
Vor wenigen Wochen nun folgte eine Verbraucherreform: Seit dem 6. April gilt in Großbritannien und Nordirland eine neue Steuer auf zuckerhaltige Soft-Drinks. Fünf oder mehr Gramm Zucker in 100 Millilitern Getränk kosten künftig 18 Pence Steuer zusätzlich (ca. 21 Cents). Bei mehr als acht Gramm werden 24 Pence fällig (ca. 28 Cents). In Irland soll ab Mai eine ähnliche Regelung in Kraft treten. Auch eine baldige Ausweitung der Abgabe auf andere Getränkearten, etwa gesüßte Milchshakes, wird diskutiert.
Hersteller reagieren mit Rezeptänderungen
Um einer höheren Besteuerung zuvorzukommen, verringerten laut Regierungsangaben bislang mehr als die Hälfte der Hersteller den Zuckergehalt ihrer betroffenen Getränke.[2] Genau das war eines der erklärten Hauptziele der Maßnahme.
Ein ähnlicher Effekt hatte sich schon in Ungarn nach Einführung einer Zuckersteuer gezeigt. Das britische Finanzministerium musste denn auch seine Prognosen korrigieren: Statt der ursprünglich geplanten Steuereinnahmen von 520 Mio. Pfund rechnet die Behörde nun nur noch mit 240 Mio. für das laufende Jahr.[3]
Der finanzielle Aspekt stand allerdings nie an erster Stelle. Die Zuckersteuer soll vielmehr Übergewicht, Diabetes und Herz-Kreislaufkrankheiten vorbeugen und auch die Zahngesundheit fördern. Im vergangenen Jahr wurden in englischen Krankenhäusern pro Tag rund 170 Kindern und Teenagern mindestens zwei Zähne entfernt.[4] Hauptursache ist der hohe Zuckerkonsum, größtenteils aus Soft-Drinks.
Zu viel oder zu wenig Reform?
Industrieverbände und Getränkehersteller kritisierten die neue Regelung massiv. Coca-Cola UK veröffentliche ein Statement in dem es u.a. hieß, es gäbe keinen verlässlichen Nachweis, dass die Besteuerung von Essen oder Getränken das Verhalten von Menschen verändert oder sie dünner mache.[5] Tatsächlich beweisen Beispiele das Gegenteil: In Mexiko fielen 2013 nach dem Inkrafttreten einer Steuer auf zuckerhaltige Getränke die Verkäufe in den folgenden zwei Jahren um zunächst 5,5% und schließlich um 9,7%, mit der stärksten Abnahme in ärmeren Haushalten.[6] Momentan erhält Coca Cola ca. 11 Gramm Zucker pro 100 Milliliter. Eine handelsübliche 0,3 Liter-Dose kommt damit auf umgerechnet 9 Teelöffel Zucker.[3] Ein neues Rezept kommt für die Firma aber nicht in Frage. Stattdessen wolle man die Flaschengröße verringern und die Preise anheben.
KritikerInnen befürchten, die neue Maßnahme könnte die Verwendung von – ebenfalls umstrittenen – Süßstoffen befördern. In Schottland ersetzte der Hersteller des populären Getränks Irn-Bru Zucker kurzerhand durch den Süßstoff Aspartam. Die künstliche Süße ist außerdem wesentlich billiger: Sie kostet nur etwa 20 % so viel wie Zucker.[3]
Und Deutschland?
Die britischen Reformen haben auch in Deutschland die Auseinandersetzung um entsprechende politische Initiativen neu befeuert. Bis 1993 hatte es eine – allerdings extrem niedrige – Zuckersteuer gegeben. Sowohl Industrie als auch das Ministerium für Ernährung und Landwirtschaft lehnen damals jedoch eine Neuauflage ab. Und auch der aktuelle Koalitionsvertrag bleibt beim Thema Zucker vage: „Für die Nationale Reduktionsstrategie für Zucker, Fett und Salz in Fertigprodukten werden wir 2018 gemeinsam mit den Beteiligten ein Konzept erarbeiten, und dies mit wissenschaftlich fundierten, verbindlichen Zielmarken und einem konkreten Zeitplan versehen.“ [2] Bundesagrarministerin Julia Klöckner lehnt Vorschriften bei Rezepturen vehement ab: „Wir definieren nicht, wie Deutschland schmeckt.“ [7] Die Organisation foodwatch veröffentlichte jüngst ihren „Coca-Cola-Report“ und wirft dem Konzern unverantwortliche Marketing- und Lobbytätigkeiten vor. Diese trügen zu steigenden Diabetes-Raten bei, speziell bei Kindern und Jugendlichen, so foodwatch. Das mediale Echo auf die Aktion war groß.
Wie zuvor in GB wurde allerdings auch im deutschen Kontext die Frage gestellt, inwieweit Maßnahmen wie die Zuckersteuer eine „Armensteuer“ darstellen. Tatsächlich ist das Gegenteil der Fall. Wie das mexikanische Beispiel zeigt, reduzieren wegen der Steuer gerade ärmere Menschen ihren Zuckerkonsum und verbessern so ihre Ernährung und damit auch ihre Gesundheit.[8]
Internationale Widerstände
Ernährung spielt eine elementare Rolle in der weltweiten Bekämpfung von Übergewicht und Diabetes. Zucker ist dabei ein Puzzleteil von vielen – aber eben ein wichtiges. Die WHO sieht sich seit Längerem der Kritik ausgesetzt, der Nahrungsmittelindustrie nicht entschieden genug die Stirn zu bieten. Eine Steuererklärung der Gates-Stiftung, größter privater Förderer der WHO, listete für 2015 Aktienanteile bei Coca-Cola im Wert von über einer halben Milliarde Dollar auf. [9]
Der Widerstand gegen eine ernsthafte Regulierung dieses Wirtschaftszweiges ist weltweit massiv. Ende 2017 widmete sich eine Reportage der New York Times dem Engagement von kolumbianischen VerbraucherschützerInnen für die striktere Besteuerung von Süßgetränken. Eine Konsequenz für die Beteiligten waren Todesdrohungen– wie der Artikel feststellte, keine Seltenheit in dem Land, aber eigentlich das bevorzugte Instrument des illegalen Drogenhandels.[10] Auch am aktuellen Beispiel Großbritannien wird sich zeigen, inwiefern gegen solche massiven Interessen nachhaltig Politik durchgesetzt werden kann. (MK)
Artikel aus dem Pharma-Brief 3/2018, S. 5
Bild: Softdrinks in supermarket © dyobmit
[1] Smyth C (2017) Britain is the fat man of Europe with 63 per cent of UK adults overweight. The Times 11 Nov. www.thetimes.co.uk/article/britain-is-the-fat-man-of-europe-as-obesity-doubles-in-two-decades-b5vx0nvsx [Zugriff 17.04.2018]
[2] Kapalschinksi C (2018) Briten erheben eine Zuckersteuer – Vorbild auch für Deutschland? Handelsblatt 6. April. www.handelsblatt.com/unternehmen/handel-konsumgueter/verbraucherschutz-briten-erheben-eine-zuckersteuer-vorbild-auch-fuer-deutschland/21146380.html [Zugriff 12.04.2018]
[3] Financial Times (2018) Sugar tax leaves bitter taste for producers. 1 April. www.ft.com/content/66cafab8-33cb-11e8-a3ae-fd3fd4564aa6 [Zugriff 14.04.2018]
[4] Guardian (2018) Dentists warn of child tooth decay crisis as extractions hit new high. 13 Jan. www.theguardian.com/society/2018/jan/13/dentists-warn-of-child-tooth-decay-crisis-as-extractions-hit-new-high [Zugriff 17.04.2018]
[5] Woods J (2016). We’re listening to consumers and taking action to reduce sugar – a tax won’t help. Coca-Cola UK 26 May. www.coca-cola.co.uk/blog/were-listening-to-consumers-and-taking-action-to-reduce-sugar-a-tax-wont-help [Zugriff 18.04.2018]
[6] Roache SA & Gostin LO (2017). The Untapped Power of Soda Taxes: Incentivizing Consumers, Generating Revenue, and Altering Corporate Behavior. In: International Journal of Health Policy and Management; Vol. 6, p 489
[7] Ärzteblatt (2018). Klöckner will der Industrie keine Rezepturen für eine gesündere Ernährung vorschreiben. www.aerzteblatt.de/nachrichten/94741/Kloeckner-will-der-Industrie-keine-Rezepturen-fuer-eine-gesuendere-Ernaehrung-vorschreiben [Zugriff 26.04.2018]
[8] Bosely S (2018) Tax sugar, alcohol and tobacco to help the poor, say experts. Guardian 4 April. www.theguardian.com/society/2018/apr/04/sin-tax-sugar-alcohol-tobacco-to-help-the-poor [Zugriff 26.04.2018]
[9] Unmüßig B (2017) Wohlwollende Alleinherrscher? www.boell.de/de/2017/11/20/milliardaere-bestimmen-globale-agenda [Zugriff 26.04.2018]
[10] Jacobs A & Richtel M (2017) She Took On Colombia’s Soda Industry. Then She Was Silenced. New York Times 13. Nov. www.nytimes.com/2017/11/13/health/colombia-soda-tax-obesity.html [Zugriff 11.04.2018]
Gesundheitsversorgung unter dem Messer
Der von Howard Waitzkin herausgegebene Band “Health Care under the Knife” ist eine harte Abrechnung mit einem profitorientierten Gesundheitswesen aus dezidiert linker Sicht.
Der Herausgeber Howard Waitzkin ist emeritierter Soziologieprofessor an der University of New Mexico und Professor für innere Medizin an der University of Illinois. Das Buch hat deshalb einen starken Fokus auf die Situation in den USA. Das gilt vor allem für die Kapitel, die sich mit dem Versorgungsalltag und den Strukturen im Gesundheitswesen, einschließlich des Zugangs zur Versorgung auseinandersetzen.
Die Analyse des medizinisch-industriellen Komplexes in den USA von Rob Burlage und Matthew Anderson mag auf den ersten Blick radikal erscheinen. Aber die präsentierten Fakten zur Macht und Gewinnorientierung von Gesundheitsversorgungs-Konzernen, ihrer engen Verflechtung mit dem Finanzsektor und akademischen Institutionen ist schon erschreckend. Wobei auch die Grenzen zwischen Universitäten und Pharmaindustrie verschwimmen. So wurde Dr. Laurie H. Glimcher von der Harvard University 2011 Dekanin der Weill Cornell Medical School. Sie hatte aber Verbindungen zu gleich zwei Firmen: Merck und Bristol-Myers. Bei letzterer war sie im Vorstand und bezog dafür 2010 eine Entlohnung in Höhe von 244.500 US$ plus Aktienoptionen im Wert von 1,4 Mio. US$.
Viele US-Amerikaner sind nicht krankenversichert und der staatliche Schutz für Menschen in Notlagen ist beschränkt. Trotzdem kommt in einem Beitrag von Waitzkin und Ida Hellander auch Obamacare - die Gesundheitsreform unter der letzten US-Regierung - nicht gut weg. Zwar hätten dadurch rund 40 Millionen BürgerInnen einen Krankenversicherungsschutz bekommen, aber die Zuzahlungen seien hoch. Außerdem profitierten die privat organisierten Versicherungen enorm.
Die AutorInnen schlagen stattdessen eine einheitliche Absicherung vor, die stark dem deutschen Modell ähnelt: Alle Menschen sind versichert und müssen nichts zuzahlen, im Krankheitsfall gibt es die gleichen Leistungen für alle.
International ausgerichtet sind andere Beiträge im Buch, so das Kapitel zur Pharmaindustrie im modernen Kapitalismus von Joel Lexchin. Es fasst die Misere, die aus der Fixierung auf Aktionäre und deren Interessen entsteht, konzise zusammen. Etwas zu knapp geraten ist der Abschnitt über die „Gesundheitskomponente des Imperialismus“. Zwar werden wichtige Akteure benannt, die negative Folgen für globale Gesundheit haben. Aber die Analysen zur Rolle der Weltbank, der Welthandelsorganisation und auch der Weltgesundheitsorganisation, die zunehmend unter dem Druck steht, vertikale Interventionen gegen einzelne Krankheiten zu propagieren, statt sich der Förderung einer umfassenden Gesundheitsversorgung zu widmen, bleiben eher oberflächlich.
Erhellend ist dagegen der Beitrag von Anne-Emanuelle Birn und Judith Richter zur Rolle des „Philanthro-Kapitalismus“ (Stichwort Gates-Stiftung), dessen Vorabdruck wir schon früher besprochen haben (Pharma-Brief 5-6/2017, S. 7).
Der Fokus auf die USA in Teilen des Buches muss nicht unbedingt ein Nachteil sein, denn hier wird besonders deutlich, welche Folgen eine Kommerzialisierung der Gesundheit hat. Das mag auch als Warnung vor ähnlichen Trends in Deutschland und anderen europäischen Ländern dienen, die auf eine Entsolidarisierung hinauslaufen und auf eine Versorgung, in der PatientInnen immer weniger im Mittelpunkt stehen. (JS)
Artikel aus dem Pharma-Brief 4-5/2018, S. 7
Bild: Cover von Waitzkin H (Hrsg.) (2018) Health Care under the Knife. Movib Beyond Capitalism for Our Health. New York: Monthly Review Press. 336 Seiten, e-book 18 US$, paperback 27 US$
Gesundheit braucht Klimaschutz!
Malaria, Dengue & Co breiten sich aus
Klimaveränderungen haben gravierende Folgen für die Gesundheit – besonders in armen Ländern. Die Pharma-Kampagne wird dieses wichtige Thema 2018 intensiv beleuchten, Forschungslücken benennen und sich für Klimaschutzziele stark machen.
Im globalen Süden sind die Auswirkungen der Erderwärmung schon jetzt deutlich zu spüren: Stürme, Überschwemmungen oder auch extreme Dürreperioden verursachen langfristige Gesundheitsprobleme. Aber auch viele Krankheiten werden durch den Klimawandel begünstigt. Die Weltgesundheitsorganisation (WHO) rechnet bis Mitte des 21. Jahrhunderts mit einem deutlichen Anstieg von Herz-Kreislauf-, Atemwegs- oder Nieren-Erkrankungen als direkter Folge des Klimawandels.[1] Massive gesundheitliche Probleme bereiten zusätzlich indirekte Effekte klimatischer Veränderung: In wärmerem Wasser können z. B. mikrobielle Keime schneller wachsen und länger überleben. Das begünstigt z. B. Wurmerkrankungen wie Bilharziose oder Durchfall-Erkrankungen wie Cholera.
Klimawandel kostet Menschenleben
Viele Erreger von Infektionskrankheiten müssen im Lauf ihrer Entwicklung im Freien überleben oder sie werden durch Zwischenwirte wie Zecken, Milben, Würmer oder Insekten übertragen. Beide Gruppen sind völlig von der Umgebungstemperatur abhängig – man bezeichnet sie als ektotherme Organismen. Bei höheren Temperaturen können sie sich schneller vermehren, entwickeln und verbreiten. Und auch die sogenannte Inkubationszeit – die Zeit zwischen der Aufnahme eines Erregers durch den Wirt und dessen Fähigkeit, den Erreger zu übertragen – verkürzt sich dramatisch.[2] Ein wärmeres Klima und stark variierende Niederschlagsmengen können zudem die geografische Ausbreitung von Krankheitsvektoren – etwa tropischer Mückenarten – stark beeinflussen. Dadurch werden Krankheiten wie Malaria oder Dengue-Fieber in Regionen zurückkehren, aus denen sie bereits erfolgreich verdrängt waren.[3] Maßnahmen zur Vektorkontrolle könnten ebenfalls ihre Wirksamkeit verlieren, warnt die WHO.
Ihren Schätzungen zufolge wird es ab 2030 jährlich 60.000 zusätzliche Todesfälle durch Malaria geben. Durchfall-Erkrankungen werden pro Jahr zusätzlich 48.000 Menschen, insbesondere Kleinkinder, das Leben kosten. Millionen zusätzlicher Krankheitsfälle werden zudem die ohnehin schwachen Gesundheitssysteme extrem fordern.[3]
Dengue-Fieber nimmt zu
Auch die Übertragungswahrscheinlichkeit von Dengue steigt in den betroffenen Regionen kontinuierlich an. Seit 1990 hat sich die Zahl der Dengue-Fälle in jedem Jahrzehnt verdoppelt. 2013 waren es weltweit 58,4 Millionen Krankheitsfälle, von denen mehr als 10.000 tödlich verliefen. Der Klimawandel ist einer der Faktoren, die erheblichen Einfluss auf diese Entwicklung haben. Beide Vektoren, Tiger- und Gelbfiebermücke, sind auch an der Übertragung anderer Krankheiten, wie Gelbfieber und Zika-Virus beteiligt, die höchstwahrscheinlich ebenso auf den Klimawandel reagieren.[4]
Ein ungebremster Klimawandel werde sämtliche Fortschritte im Bereich öffentliche Gesundheit zunichtemachen, die in den vergangenen 50 Jahren erreicht wurden, warnt die Fachzeitschrift The Lancet.4 Andererseits könnten umfassende und ganzheitliche klimagerechte Handlungsstrategien die größte Gesundheitschance des 21. Jahrhunderts darstellen. Denn sie fördern zugleich einen gesünderen Lebensstil.
Geplante Aktionen
Effektive Klimapolitik ist aus vielerlei Hinsicht überlebenswichtig für unseren Planeten. Sie ist aber auch eine Frage der Gerechtigkeit und des Menschenrechts auf Gesundheit. Unsere Theatertournee wird im September über diese Zusammenhänge informieren. Begleitend gibt es neue Bildungsmaterialien, etwa großformatige Infotafeln, die bei Veranstaltungen eingesetzt werden können sowie Online-Materialien. Ein Pharma-Brief Spezial zum Thema Klimawandel und globale Gesundheit erscheint im Herbst. (CJ)
Artikel aus dem Pharma-Brief 2/2018, S.1
[1] WHO (2015) Climate and Health Country Profiles 2015 – A Global Overview. http://apps.who.int/iris/bitstream/10665/208855/1/WHO_FWC_PHE_EPE_15.01_eng.pdf?ua=1 [Zugriff 26. 2. 18]
[2] Hutter, Moshammer, Wallner (2017) Klimawandel und Gesundheit. Auswirkungen. Risiken. Perspektiven, Wien: Manz, S.71ff.
[3] WHO (2018) Climate change and health. Verfügbar unter: www.who.int/mediacentre/factsheets/fs266/en/ [Zugriff 22. 2. 2018]
[4] Watts N et al. (2017) The Lancet Countdown on health and climate change: from 25 years of inaction to a global transformation for public health. The Lancet; 391, p 9-10
EU-HTA Update 2
Europäische Nutzenbewertung im Fluss
Wir berichteten wiederholt über die Diskussion um den Gesetzesvorschlag der EU-Kommission zum EU-HTA.[1],[2] Was gibt es Neues?
Jetzt liegen auch die konsentierten Kompromissvorschläge des federführenden Ausschusses für Umweltfragen, öffentliche Gesundheit und Lebensmittel (ENVI) des EU-Parlaments vor.[3] Gegenüber dem Kommissionsvorschlag gibt es einige deutliche Verbesserungen. Wichtig ist, dass die HTA-Bewertung nicht mehr parallel mit dem Zulassungsprozess durchgeführt werden soll, sondern im Anschluss. Die Berichterstatterin des ENVI-Ausschusses, Soledad Cabezón Ruiz, betonte, dass eine schnelle Marktverfügbarkeit nicht das erste Kriterium sein könne: „Für uns ist es sehr wichtig, dass die Qualität im Vordergrund steht, […] nicht die Geschwindigkeit“.[4] Deshalb wurde der Anspruch, dass die Bewertung des Nutzens zum Zeitpunkt der Zulassungsentscheidung abgeschlossen sein soll, gekippt.
Beschlüsse über die Bewertung eines Arzneimittels oder Medizinprodukts sollen im Streitfalle nur mit einer 2/3 Mehrheit der Mitgliedsstaaten verabschiedet werden können. Vorgesehen war eine einfache Mehrheit.
Transparenz
Alle Berichte und Schlussfolgerungen aus dem HTA-Verfahren sollen in eine öffentliche Datenbank eingestellt werden. Dadurch werden mehr Informationen zugänglich sein als das gegenwärtig bei der europäischen Zulassungsbehörde EMA der Fall ist. Schwärzungen von vertraulichen Daten soll es aber nach wie vor geben.
Zwang aufgeweicht
Eine verpflichtende Übernahme der EU-HTA-Bewertungen in allen Mitgliedsstaaten wurde aufgeweicht. Es heißt jetzt nur noch „die Mitgliedsstaaten sollen die Ergebnisse der gemeinsamen klinischen Bewertung berücksichtigen.“ Ergänzende nationale Bewertungen sollen möglich sein, um spezifische klinische Umstände zu berücksichtigen oder jede andere Frage zu beantworten. Allerdings soll das kein Freibrief sein, generell auf eine nationale HTA-Bewertung zu verzichten.
Außerdem wurde klargestellt, dass die europäische Bewertung die klinischen Fakten ausführlich und transparent darstellen soll, die letztendliche Wertung des klinischen Nutzens aber nationale Angelegenheit bleibt.
Der neue Entwurf ist schon nah an dem Kompromissvorschlag, den Frankreich und Deutschland in die Debatte eingebracht hatten. Dem Vernehmen nach soll sich inzwischen die Mehrheit der GesundheitsministerInnen gegen eine verpflichtende Übernahme der europäischen Bewertung ausgesprochen haben. Das EU-Parlament wird voraussichtlich im Oktober endgültig über die EU-Verordnung zu HTA abstimmen. Danach ist der Ministerrat am Zug.
HTA = Zugang?
Viele EU-ParlamentarierInnen sind der Ansicht, dass eine verpflichtende europäische HTA-Bewertung den Zugang zu neuen Arzneimitteln verbessern würde. Das ist ein gravierendes Missverständnis. Denn viele neue Produkte werden trotz EU-Zulassung von den Herstellern in ärmeren Mitgliedsstaaten erst später oder gar nicht auf den Markt gebracht. Auch hohe Preise werden weiterhin ein Zugangshindernis bleiben. Denn es ist unstrittig, dass jeder HTA-Bewertung eine Kosten-Nutzen-Bewertung folgen kann. Bisherige Erfahrungen aus Mitgliedsstaaten zeigen, dass astronomische Preise bei bescheidenem Zusatznutzen oft zum Ausschluss aus der Erstattung führen.
Beide Themen geht die österreichische Ratspräsidentschaft an. In einem Diskussionspapier für ein informelles GesundheitsministerInnen-Treffen wird vorgeschlagen, den Artikel 14 der EU-Verordnung 726/2004 strenger auszulegen.[5] Bislang sah es die EU-Kommission als ausreichend an, wenn der Hersteller sein neues Produkt mindestens in einem Mitgliedsstaat auf den Markt brachte. Künftig könnte er seine Zulassung verlieren, wenn er sein Medikament nicht in allen EU-Staaten anbietet.
Auch zum Thema Evidenz und Preise hat das Alpenland einiges im Köcher. Das Diskussionspapier fordert, in klinischen Studien nur noch gegen die Standardtherapie zu testen und nicht mehr gegen Placebo. Dabei sollen in patientenrelevanten Endpunkten Vorteile gezeigt werden. Außerdem wollen die Österreicher einen früheren und vollständigeren Zugang zu allen Studiendaten. Das mache verlässliche HTA-Bewertungen überhaupt erst möglich. Sprengstoff enthält auch die Forderung, die Forschungskosten offenzulegen – einschließlich der öffentlichen Förderung. (JS)
Artikel aus dem Pharma-Brief 7/2018, S.6
[1] Pharma-Brief (2018) Wunschkonzert für Hersteller. Nr. 3, S. 1
[2] Pharma-Brief (2018) Zwischen Kommerz und Transparenz. Nr. 6, S. 5
[3] Die Vorschläge aus dem Vorbereitungsdokument (und acht weitere) wurden alle angenommen: www.europarl.europa.eu/meetdocs/2014_2019/plmrep/COMMITTEES/ENVI/DV/2018/09-13/2018-09-12_Final-COMP_HTA-ENVI-ALL_EN.pdf
[4] Wheaton S (2018) Parliament negotiators reach deal on HTA position. Politico. 7 Sept
[5] Austria (2018) Informal Meeting of Health Ministers on 10 and 11 September 2018. Regulatory and policy-related challenges in securing supply of centrally authorised medicines
Die stille Epidemie
Diabetes im Fokus
425 Millionen Menschen leiden weltweit unter Diabetes - die meisten davon in Ländern geringen oder mittleren Einkommens. Doch gerade dort haben die PatientInnen wenig Hoffnung auf eine gute Versorgung: Insulin ist in den meisten armen Ländern schlecht verfügbar und die Erkrankung treibt Betroffene und deren Familien häufig in die Armut. Mit einem neuen E-learning-Kurs will die Pharma-Kampagne auf diese Probleme aufmerksam machen. Die Online-Materialien sollen Ende des Jahres erscheinen.
„Zigarettenhersteller dürfen Olympia nicht sponsern. Warum darf es Coca-Cola?“,[1] so titelte ein im Guardian erschienener Gastbeitrag zur Eröffnung der Winterspiele in Pyeongchang/Südkorea. Eine berechtigte Frage, denn Werbung für Tabak ist im Rahmen der Spiele zwar seit 1988 verboten - nicht jedoch die für Fast Food-Produkte oder zuckerhaltige Getränke. Die sind aber nicht minder schädlich, fördern sie doch massiv Übergewicht und Folgeerkrankungen wie Diabetes. Trotzdem besteht die unsportliche Allianz von Coca-Cola mit dem Internationalen Olympischen Komitee (IOC) bereits seit 1928 und soll noch bis mindestens 2020 fortgesetzt werden.[2]
Das Beispiel zeigt einmal mehr die vorherrschende Ignoranz gegenüber Diabetes als wachsendem globalem Gesundheitsproblem. In keiner anderen WHO-Region leben mehr Diabetes-PatientInnen als in der bevölkerungsreichen Westpazifik-Region, zu der auch der diesjährige Gastgeber der Winterspiele zählt.[3] Auch Südkorea selbst verzeichnet steigende Prävalenzraten.[4]
Komplexe Ursachen
So fragmentiert die Datenlage besonders für ärmere Länder noch ist, zeigt sich ein deutlicher Trend: Laut Schätzungen des „Atlas“ der International Diabetes Foundation (IDF) sind mittlerweile 425 Millionen Menschen weltweit an Diabetes erkrankt, davon leben 79% in Ländern geringen oder mittleren Einkommens.[5] Generell haben Bevölkerungswachstum und steigende Lebenserwartung Einfluss auf die hohen Zahlen. Doch das erklärt nur zum Teil, warum sich die globale Prävalenz zwischen 1980 und 2014 laut Weltgesundheitsorganisation (WHO) fast verdoppelt hat (von 4,7% auf 8,5%).[6]
Tabakkonsum und Übergewicht
Die Ursachen für diese Dynamik sind komplex, denn die Entstehung von Typ 2 Diabetes, der häufigsten Form der Erkrankung, hängt von vielen Faktoren ab: Neben genetischen Voraussetzungen hat u.a. Tabakkonsum großen Einfluss. Eine zentrale Rolle spielen zudem Übergewicht und Adipositas.[7] Ursächlich dafür ist wiederum der starke Wandel des Lebensstils aufgrund massiver wirtschaftlicher und sozialer Umbrüche in vielen Gesellschaften des globalen Südens. Geringere körperliche Aktivität und veränderte Ernährungsmuster sind oft eine Folge beschleunigter Urbanisierung. Sie treffen auf wenig vorbereitete und schlecht ausgestattete Versorgungssysteme, die bereits bei der Behandlung der vorherrschenden Infektionskrankheiten häufig an ihre Grenzen stoßen.[8]
Engpässe bei Diagnose und Behandlung
In armen Ländern hapert es gewaltig bei der Diagnose und Therapie von Diabetes. Insulin - ein unerlässliches Präparat für Millionen PatientInnen weltweit - ist nur in knapp einem Viertel der Länder mit niedrigem Einkommen generell verfügbar.[9] Fehlende Behandlung schädigt aber wiederum Herz, Blutgefäße, Nieren, Augen und Nerven und führt häufig zu Invalidität. Amputationen der unteren Extremitäten sind bei DiabetikerInnen z. B. 10-20 mal so häufig wie bei Gesunden. Aber Diabetes ist auch verantwortlich für jährlich 1,5 Millionen Todesfälle. Zusätzlich begünstigt ein hoher Blutzucker Herz-Kreislauf-Erkrankungen.
Der Markt für Insulin wird von lediglich drei Anbietern dominiert (Eli Lilly, Novo Nordisk & Sanofi). Auch diese Marktkonzentration begünstigt hohe Preise und sorgt für Engpässe. Eine Studie von Health Action International stellte 2017 fest, dass Insulin in vielen Ländern für PatientInnen schwer zu finanzieren ist. Zudem ist erstaunlicherweise der Preis von älteren Präparaten nicht spürbar gefallen wie es in der Regel der Fall ist.
Preis bleibt hoch
“Der globale Einkaufspreis (…) scheint über die Zeit hinweg unverändert geblieben zu sein - ganz anders als bei anderen NCD-Medikamenten oder HIV-Therapien.”[10] Aber auch an schlichten Blutzucker-Teststreifen oder Injektionszubehör mangelt es in vielen ressourcenschwachen Regionen. Zudem ist die Fallfindung miserabel: Die IDF schätzt, dass die Hälfte aller Erkrankten zwischen 20 und 79 Jahren nie eine entsprechende Diagnose erhalten hat.[5] Und auch die Prävention kommt zu kurz. Dabei ließen sich dafür oftmals bestehende Versorgungsstrukturen nutzen.[11]
Wirksame Konzepte gefragt
Um der zunehmenden Verbreitung von Diabetes und anderer nicht übertragbarer Krankheiten (NCDs) Rechnung zu tragen, berief die WHO im Februar eine unabhängige Kommission: Die Independent Global High-level Commission on NCDs soll Strategien und Maßnahmen entwickeln, die geeignet sind, um NCDs wirksam einzudämmen und die Sterberaten zu senken.[12] Gelingt das nicht, werden letztlich auch die nachhaltigen Entwicklungsziele scheitern, deren Umsetzung sich die Vereinten Nationen bis 2030 vorgenommen haben. Diesen Realitäten muss auch die Entwicklungszusammenarbeit und humanitäre Hilfe deutscher NROs verstärkt Rechnung tragen.
Neuer E-learning-Kurs
Ein neuer Online-Kurs der BUKO Pharma-Kampagne will MitarbeiterInnen der Entwicklungszusammenarbeit auf diese Herausforderungen vorbereiten. Die E-Learning-Module „Diabetes – die stille Epidemie“ werden derzeit entwickelt und sollen Ende des Jahres auf unserer Website kostenlos zur Verfügung stehen. Ziel ist es, MitarbeiterInnen in Gesundheitsprojekten für die Probleme zu sensibilisieren und damit die Versorgung der PatientInnen, aber auch die Prävention zu verbessern.
Neben medizinischen und epidemiologischen Informationen sowie praktischen Handlungsempfehlungen wird der Kurs eine klinisch-pharmakologische Bewertung häufig eingesetzter Antidiabetika beinhalten. (MK)
Artikel aus dem Pharma-Brief 2/2018, S. 3
Bild © Brian Finney
[1] The Guardian (2018) Cigarette companies don´t sponsor the Olympics. Why does Coca-Cola?. www.theguardian.com/commentisfree/2018/feb/10/coca-cola-mcdonalds-sponsor-olympics [Zugriff 13. 2. 2018]
[2] Coca-Cola Journey (2016) Die gemeinsame Geschichte von Coca-Cola und den Olympischen Spielen seit 1928. 19.8.2016 https://de.coca-cola.ch/stories/die-gemeinsame-geschichte-von-coca-cola-und-den-olympischen-spielen-seit-1928 [Zugriff 21. 2. 2018]
[3] Nanditha A et al. (2016) Diabetes in Asia and the Pacific: Implications for the Global Epidemic. In: Diabetes Care; Vol. 39, p 472-485
[4] Noh J et al. (2017) Trends in the pervasiveness of type 2 diabetes, impaired fasting glucose and co-morbidities during an 8-year-follow-up of nationwide Korean population, In: Scientific Reports; 7, p 1-7, S. 5
[5] IDF (2017) IDF Diabetes Atlas 2017. www.idf.org/e-library/epidemiology-research/diabetes-atlas/134-idf-diabetes-atlas-8th-edition.html. S. 43 [Zugriff 13. 2. 2018]
[6] WHO (2017) Diabetes Fact sheet. www.who.int/mediacentre/factsheets/fs312/en [Zugriff 13. 2. 2018]
[7] Hu, Frank B (2011) Globalization of Diabetes. The role of diet, lifestlye, and genes. Diabetes Care; Vol. 34, p 1249-1257.
[8] NYT (2018) In Kenya and Across Africa, an Unexpected Epidemic: Obesity. www.nytimes.com/2018/01/.../kenya-obesity-diabetes.html [Zugriff 11. 2. 2018]
[9] Chan M (2016) Opening remarks on World Health Day and the launch of the WHO Global report on diabetes. Geneva, Switzerland, 7 April 2016
[10] HAI (2017) Access to insulin: Current challenges and constraints, Amsterdam, S. 23
[11] WHO (2016) The mysteries of type 2 diabetes in developing countries. In: Bulletin of the World Health Organization, S. 242. www.who.int/bulletin/volumes/94/4/16-030416.pdf [Zugriff 5. 2. 2018]
[12] WHO (2018) WHO Independent High-level Commission on NCDs. www.who.int/ncds/governance/high-level-commission/en/ [Zugriff 14. 2. 2018]
Die Schattenseiten des Vitamin D-Papstes
Über die Erfindung eines Gesundheitsproblems
Niemand hat die Verwendung von Vitamin D zur Verhinderung aller möglichen Erkrankungen mehr propagiert als der US-amerikanische Arzt Michael Holick. Seine weitgehend evidenzfreien Empfehlungen setzten sich nicht nur landesweit durch, sondern haben auch international zum Vitamin D-Hype beigetragen. Ein Artikel in der New York Times gibt Aufschluss darüber, wie es dazu kam.[1]
Holick hat das Publikum mit zahllosen populärwissenschaftlichen Artikeln und einem Buch überzeugt.[2] Er verstieg sich sogar zu der These, dass die Dinosaurier unter anderem wegen Vitamin D-Mangels ausgestorben seien. Der Umsatz von Vitamin D-Ergänzungsmitteln in den USA hat sich in einer Dekade verneunfacht.
Der Wissenschaftler hat aber auch dafür gesorgt, dass ÄrztInnen fleißig auf Vitamin D-Mangel testeten, 2016 wurden über zehn Millionen Medicare [3] PatientInnen darauf untersucht – das sind gut fünfmal so viele wie 2007. Wie es dazu kam? 2011 hatte die angesehene National Academy of Medicine (heute Institute of Medicine) einen langen Bericht über den Vitamin D-Mangel veröffentlicht. Die Quintessenz: Die meisten Menschen bekommen über Nahrung und Sonnenlicht reichlich von dem Vitamin und eine Testung ist nur bei Menschen mit hohem Risiko – wie zum Beispiel bei Osteoporose – sinnvoll.
Leidlinie?
Nur wenige Monate später leitete Dr. Holick eine Arbeitsgruppe der Endocrine Society zu Vitamin D. In dieser Fachgesellschaft sind die meisten Spezialisten organsiert. Ihre Leitlinien werden von zahllosen Krankenhäusern, ÄrztInnen und kommerziellen Labors befolgt. Die Endocrine Society akzeptierte Holicks Urteil, dass „Vitamin D-Mangel in allen Altersgruppen sehr verbreitet ist“. Anders als die Empfehlung der National Academy, die 20 Nanogramm als ausreichend erachtete, setzte die Endocrine Society den Grenzwert auf 30 Nanogramm hoch. Die Leitlinie machte damit nach Aussage von Dr. Clifford Rosen, Co-Autor des Berichts der National Academy, 80% der US-Bevölkerung zu potenziell Kranken. „Wir sehen, dass ständig Leute getestet und anschließend behandelt werden. Die Basis dafür ist eine gute Portion Wunschdenken, dass man ein Nahrungsergänzungsmittel einnehmen kann, um dadurch gesünder zu werden.“[1]
Gekauft?
Mit den Tests lässt sich eine Menge Geld verdienen, sie kosten in den USA zwischen 40 und 225 US$. Auch Dr. Holick verdient an diesem Geschäft. Er bekommt seit 40 Jahren Geld von Quest, einem führenden Anbieter von Vitamin D-Tests. Gegenwärtig erhält er 1.000 US$ im Monat. Seiner Meinung nach beeinflusst das sein Urteil nicht: „Ich bekomme nicht mehr, ob einer oder eine Million Tests verkauft werden.“[1] Die Branche dankt ihm sein Engagement jedenfalls mit fürstlichen Honoraren. Von 2013 bis 2017 erhielt Holick insgesamt fast 163.000 US$ von Pharmafirmen, darunter auch von mehreren Vitamin D-Herstellern und zwei Firmen, die die dazu passenden Tests verkaufen.
Sonnenstudio statt Sonnenlicht
Holick propagiert, sich der Sonne auszusetzen, und das möglichst viel, da er ja sehr hohe Vitamin D-Spiegel im Körper für wichtig hält. Zwar ist Sonnenlicht für die körpereigene Vitamin D-Produktion notwendig, aber über das Ausmaß kann man wegen des Hautkrebsrisikos streiten. Wirklich fragwürdig wurde es, als er Sonnenstudios als Vitamin D-Quelle empfahl. Eine gemeinnützige Lobbyeinrichtung der Sonnenbank-Industrie spendete der Uni Boston von 2004 bis 2006 150.000 US$, Verwendungszweck: Die Forschung von Dr. Holick.
Holicks Werbefeldzug für Vitamin D wurde von der Wellness-Industrie begierig aufgegriffen. Dr. Oz, der eine populäre Website zu Gesundheit anbietet, schreibt dem Vitamin Wunderwirkungen zu. Es helfe gegen Herzkrankheiten, Depressionen, Vergesslichkeit und Krebs. Auch die bekannte US-Talkshow-Moderatorin und Schauspielerin Oprah Winfrey wirkt als Propagandistin: „Wenn du deinen Vitamin D-Level kennst kann das dein Leben retten.“
Gegen diesen massiven Propaganda-Wirbel für Vitamin D hatten es die Empfehlungen der National Academy of Medicine von 2011 schwer. Aber langsam dreht sich der Wind.
Sinkender Stern
Heute könnte Holick nicht mehr Vorsitzender der Leitliniengruppe bei der Endocrine Society werden, denn die Gesellschaft hat ihre Regeln für Interessenkonflikte verschärft.
Bereits 2015 warnte eine von den US-National Institutes of Health einberufene ExpertInnenkonferenz vor ernsten Gesundheitsschäden durch zu hohe Vitamin-D Dosen. Schon ein Spiegel von 50 Nanogramm wurde als möglicherweise gefährlich bezeichnet.[4] Diese Menge liegt im Bereich der noch gültigen Empfehlung der Endocrine Society-Leitlinie.
Immer mehr Forschung zeigt, dass an den Versprechen des Vitamin D-Papstes nichts dran ist. Bereits 2014 machte eine große Metaanalyse deutlich, wie schwach die Datenlage für viele behauptete Vorteile des Vitamins ist.[5] Im November diesen Jahres zeigte eine gut gemachte große randomisierte Studie, dass das Vitamin weder Krebs noch Herz-Kreislauferkrankungen verhindern kann.[6]
Vermutlich ist es eher umgekehrt, gebrechliche Menschen sind nicht krank, weil sie zu wenig Vitamin D haben, sondern sie haben zu wenig davon, weil sie kaum mehr nach draußen kommen. (JS)
Artikel aus dem Pharma-Brief 10/2018, S. 3
[1] Szabo L (2018) Vitamin D, the sunshine supplement, has shadow money behind it. New York Times 18 Aug.
[2] Holick M (2011) The vitamin D solution. New York: Plume
[3] Durch das staatliche Medicare-Programm sind 58 Millionen BürgerInnen in den USA (teilweise) gegen Krankheit abgesichert. Die meisten sind über 65 Jahre alt. Medicare (2018) Annual Report of the Medicare Trustees (for the year 2017), June 8
[4] Taylor C et al. (2015) Questions About Vitamin D for Primary Care Practice: Input From an NIH Conference . The American Journal of Medicine; 128, p 1167
[5] Theodoratou E et al. (2018) Vitamin D and multiple health outcomes: umbrella review of systematic reviews and meta-analyses of observational studies and randomised trials. BMJ;348, p g2035
[6] Manson JE et al. (2018) Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. NEJM. DOI: 10.1056/NEJMoa1809944
Der Dollar rollt langsam
Erstmals seit 2012 mehr Geld für vernachlässigte Krankheiten
Wieviel Geld wird in die Forschung und Entwicklung zu vernachlässigten Krankheiten investiert? Der Bericht G-FINDER zeigt für 2016 eine spannende Tendenz. Auch wenn die USA mit Abstand der wichtigste Geldgeber bleibt, legen Indien und Südafrika verhältnismäßig deutlich zu.
Der G-FINDER ist eine Institution.[1] Ein Team um das australische Institut Policy Cures Research stellt jährlich umfangreiche Daten zusammen, wer wieviel Geld in die Erforschung vernachlässigter Krankheiten investiert. Erfasst werden derzeit 33 Erkrankungen. Im Jahr 2016 lagen die weltweiten Aufwendungen für die Erforschung von Impfstoffen, Medikamenten und Diagnostik bei 3,26 Mrd. US$. Erstmals seit 2012 stiegen die Investitionen wieder an, und zwar um 3,4%. Die meisten Gelder gehen aber in die „großen Drei“ Malaria, HIV und Tuberkulose. Dort sind ebenfalls Steigerungen zu verzeichnen, mit Ausnahme von Tuberkulose (minus 6,8%).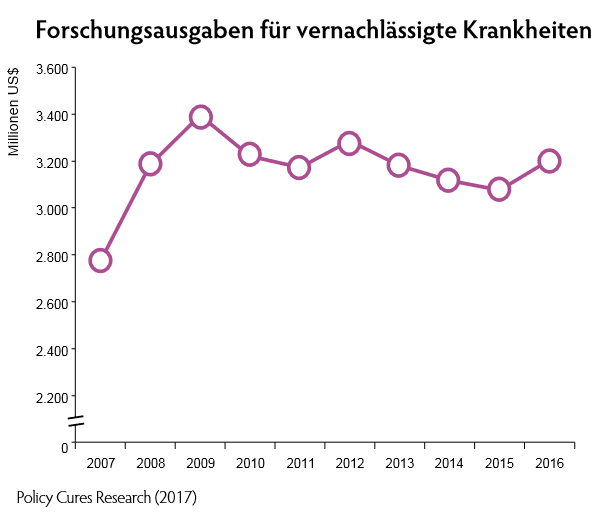
Die übrigen Ausgaben verteilen sich unter anderem auf Wurmerkrankungen, Dengue, Salmonellen-Infektionen, aber auch bestimmte Genotypen von Hepatitis C, die vor allem ärmere Länder betreffen. Für einige Regionen wurden auch bestimmte Auslöser von Lungenentzündung (Streptococcus pneumoniae) und Hirnhautentzündung (Neisseria meningitidis) in die Statistik einbezogen.
Platz 1: USA
Öffentliche Finanzierung spielt mit 64% nach wie vor die wichtigste Rolle, gefolgt von philanthropischen Stiftungen (21%) und der Industrie (16%).[2] Bei den staatlichen Geldgebern ist die Vorreiterrolle der USA ungeschlagen: Sie bringen mit 1,49 Mrd. US$ dreimal so viel Geld auf wie alle anderen Regierungen der Welt.
Mit großem Abstand folgt Großbritannien auf Platz zwei (101 Mio. US$), gefolgt von der EU (77 Mio. US$), Indien (50 Mio. US$) und Frankreich (47 Mio. US$). Deutschland liegt mit einer Fördersumme von 43 Mio. US$ eher im Mittelfeld (Platz 6). Andere bedeutsame Geldgeber sind die Niederlande, Australien, Brasilien, die Schweiz, Japan und Schweden.
Rolle von Wachstumsstaaten
Ein wichtiges Signal setzen drei Schwellenländer: Brasilien, Südafrika und Indien tragen inzwischen 84 Mio. US$ zum Gesamttopf bei und haben damit deutlich zugelegt. Die Förderung der EU ist 2016 stark eingebrochen, was aber vor allem daran liegt, dass das Programm EDCTP zur Förderung klinischer Studien 2016 80% weniger Geld ausgeben konnte als 2015, wo es hohe außerplanmäßige Ausschüttungen gegeben hatte.
Deutschland könnte mehr
Anschaulicher werden die Zahlen, wenn man die Ausgaben in Relation zum Bruttoinlandsprodukt (BIP) betrachtet. Wenn ein Wert von 10 bedeutet, dass ein Land 0,01% seines BIP für vernachlässigte Krankheiten ausgibt, dann liegt die USA mit 8,0 nach wie vor an der Spitze. Großbritannien folgt mit Abstand bei 3,8. Dann kommen schon Südafrika mit 3,2 und Indien mit 2,2. Deutschland folgt erst bei 1,2. Es ist also noch viel Luft nach oben.
Gates
Hinter den philanthropischen Geldgebern stecken zwei altbekannte Namen: die Gates Foundation und der Wellcome Trust. Diese spielen eine wichtige Rolle für Produktentwicklung und klinische Studien, wogegen die öffentlichen Geldgeber (mit Ausnahme der HIV-Vakzineforschung) hauptsächlich Grundlagenforschung fördern.
Industrie
Das meiste Firmengeld stammt von multinationalen Konzernen. Deren Investitionen stagnieren allerdings seit einigen Jahren. Dass bei der Industrie dennoch ein Wachstum verzeichnet werden kann, ist fast ausschließlich kleinen und mittleren Unternehmen zu verdanken. Vor allem in Ländern mit mittlerem Einkommen haben diese Unternehmen um 30% zugelegt, besonders im Bereich klinische Studien. (CW)
Artikel aus dem Pharma-Brief 2/2018
[1] Policy Cures Research (2017) Neglected disease research and development: Reflecting on a decade of global investment. www.policycuresresearch.org/g-finder
[2] rundungsbedingte Fehler
Das Ende von HIV in Sicht?
Das „Aktionsbündnis gegen AIDS“ zieht nach 15 Jahren Bilanz
Vor einigen Jahren schien es kaum denkbar, dass einmal weltweit 18 Millionen Menschen mit einer HIV-Therapie versorgt werden könnten. In vielen Ländern des globalen Südens ist heute eine gute Versorgung Realität. Das ist auch einem starken zivilgesellschaftlichen Engagement in Deutschland zu verdanken. Bei einer Konferenz anlässlich seines 15-jährigen Jubiläums richtete das „Aktionsbündnis gegen AIDS“ den Blick aber auch nach vorne: Welche Hürden sind noch zu nehmen, um ein Ende von Aids zu erreichen?
Das Ziel ist klar: Auch wenn sich HIV-Infektionen nicht vollständig vermeiden lassen, soll niemand an Aids sterben müssen. Das Ende von Aids wurde 2014 von UNAIDS in die Formel 90-90-90 gefasst:[1] [2] 90 Prozent aller HIV-Infizierten sollen ihren Status kennen; 90 Prozent der HIV-Positiven sollen antiretrovirale Behandlung erhalten; bei 90 Prozent der Behandelten soll die Viruslast unter der Nachweisgrenze liegen. Dieses Ziel soll im Jahr 2020 erreicht sein.
Das deutsche Aktionsbündnis gegen AIDS will dazu einen Beitrag leisten. Das als „Aidskampagne“ bezeichnete Bündnis wurde 2002 gegründet und bringt mittlerweile über 300 Gruppen aus Kirchen, politischen Organisationen und lokalen Aids-Hilfe Gruppen zusammen. Auch die Pharma-Kampagne ist von Anfang an dabei. Unter dem Motto „Leben ist ein Menschenrecht“ setzt sich die Aidskampagne für Therapie und gegen Diskriminierung ein.
Was das im Einzelnen bedeutet, wurde im Verlauf der Veranstaltung sehr deutlich. Geladen waren nationale und internationale Gäste, die die Herausforderungen für die Zukunft aus ihrem Kontext schilderten.
Problem Diskriminierung
Wie stark die Diskriminierung von Menschen mit HIV die grundlegenden Menschenrechte bedroht, beschrieb der russische Journalist und Aktivist Alexander Delphinov am Beispiel von DrogenkonsumentInnen. In Russland sind derzeit 700.000 Menschen im Gefängnis, davon ein Drittel DrogengebraucherInnen. Deren Leben sei auch außerhalb der Gefängnisse von Angst vor Gewalt und Polizeiwillkür geprägt. „DrogenkonsumentInnen werden wie Tiere behandelt, und Tiere haben keine Menschenrechte“. Selbst gegenüber ÄrztInnen gäbe es großes Misstrauen, da diese oft Informationen über den HIV-Status und Drogenkonsum an die Polizei weiterleiten. In dieser Situation sei Solidarität lebensrettend: Menschen mit HIV, DrogenkonsumentInnen und SexarbeiterInnen müssten sich gegenseitig stützen.
Die gefährliche Situation von Homosexuellen in Nordafrika schilderte Ahmed Awadalla von der Deutschen Aidshilfe. Obwohl Homosexualität in Ägypten schon länger kriminalisiert ist, habe es viele Jahre Präventionsarbeit, Testberatung, Sexualaufklärung in Schulen und bei Familien gegeben. 2011 gab es mit dem arabischen Frühling Hoffnung auf Besserung, dann trat aber 2013 die Kehrtwende ein. Hunderte Menschen sitzen nun wegen ihrer sexuellen Orientierung im Gefängnis. Die Polizei nutzt Dating-Apps wie Tinder, um Homosexuelle zu identifizieren und zu verfolgen. Die Betroffenen leben in permanenter Angst. Auch die Bereitschaft, sich auf HIV testen zu lassen, ist gering, da ein positives Ergebnis als Beweis für Homosexualität gewertet wird. Homosexualität ist deshalb als Fluchtursache nicht zu unterschätzen, aber die Asyl-Anerkennung sei schwierig.
Ist die Situation in Deutschland besser? Die Pfarrerin Dorothea Strauß, Leiterin von „Kirche positHIV“, stellte klar, dass auch die Kirchen in den 80er Jahren nur schwer mit Homosexualität umgehen konnten. Im Laufe der Jahre habe sich zum Glück viel getan. Das Thema Homosexualität sei jetzt eine große Herausforderung für den Austausch mit afrikanischen Partnerkirchen. Dennoch ist Stigmatisierung in Deutschland immer noch ein Problem. Silke Klumb von der Deutschen Aidshilfe verdeutlichte das anhand des „Stigma-Index“, der durch Umfragen die Erfahrungen mit Ausgrenzung erfasst.[3] In Deutschland hat jedeR Dritte negative Erfahrungen gemacht, wenn er oder sie sich mit dem eigenen HIV-Status outete. Das schafft Risiken: Wenn Menschen die Erfahrung von Diskriminierung machen, hören sie auf über HIV zu sprechen. Die Krankheit wird tabuisiert und neue Infektionen sind die Folge. Und auch Angst macht krank.
Zugang zur Behandlung
Die Verfügbarkeit von Medikamenten hat sich in den letzten Jahren stetig verbessert. Die Standard Einstiegs-Therapie (Firstline-Treatment) ist in vielen ärmeren Ländern bereits für 100 US$ pro Person und Jahr erhältlich. Teurer ist die zweite Behandlungslinie (Secondline-Treatment) mit 300 US$. Die Thirdline-Therapie kostet dann bereits über 1.000 US$. Eine wichtige Rolle für den Zugang zu kostengünstigen Therapien spielt der Medicines Patent Pool. Er wurde 2010 gegründet und schließt freiwillige Vereinbarungen mit Patentinhabern ab, in den meisten Fällen Pharmaunternehmen. Die Verträge erlauben eine einfache Vermittlung von Lizenzen an Generikahersteller und ermöglichen so Produktion und Vermarktung günstiger HIV-Medikamente. Nach anfänglicher Mühe, die Pharmaunternehmen zur Mitarbeit zu bewegen, wurde das Modell so erfolgreich, dass es 2015 auf TB und Hepatitis C erweitert wurde. Erika Dueñas vom Medicines Patent Pool berichtete, dass derzeit geprüft werde, ob eine Ausweitung auf alle patentgeschützten Arzneimittel in der Liste unentbehrlicher Medikamente der WHO möglich ist.
Einen Einblick in den Behandlungsalltag gab Schwester Melania, die sich in einem Distriktkrankenhaus in Zimbabwe vor allem um junge Menschen mit HIV kümmert. Antiretrovirale Medikamente werden von der Regierung gestellt. Für opportunistische Infektionen – also Folgen der HIV-Infektion – wie Pilzbefall, Lungenentzündung oder bestimmte Krebsarten, fehlen jedoch häufig die Behandlungsmöglichkeiten.
Schwester Melania verdeutlichte das Problem der Stigmatisierung im Zusammenhang mit HIV-Tests: Männer lassen sich häufig scheiden, wenn ihre Frau ein positives Testergebnis hat – lassen sich aber selbst nicht testen. Wenn die Frau wieder heiratet, verschweigt sie oft ihren HIV-Status aus Angst, wieder verlassen zu werden. Auch wenn junge Frauen vor dem ersten Geschlechtsverkehr bereits positiv getestet werden, sorgt das für persönliche und soziale Probleme. Hier hilft Aufklärung und Bildung, um klar zu machen, dass die Infektion häufig schon bei der Geburt oder durch Stillen übertragen wird.
Welche Erfolge das Engagement der Zivilgesellschaft bringen kann, zeigt das Beispiel Ukraine. Ein Netzwerk von Menschen mit HIV startete eine Kampagne, damit die Regierung die Therapiekosten für Hepatitis C senkt. Von 45 Millionen EinwohnerInnen haben schätzungsweise 3,5 Mio. eine Hepatitis-Infektion, viele davon sind auch HIV-positiv. Eine Medienkampagne, Theateraktionen und Demonstrationen bauten Druck auf und erreichten das Ziel: Der Preis sank von 15.000 US$ (2014) auf 900 US$ (2017).
Wer soll’s bezahlen?
Am Schluss der Veranstaltung stand eine zentrale Frage: Wie sind die notwendigen Maßnahmen zu finanzieren? Bis 2020 sollen schließlich 90% aller HIV-Positiven weltweit eine Therapie erhalten, das sind 30 Millionen Menschen.[4] Eine zentrale Rolle spielt seit etlichen Jahren der Global Fund to Fight AIDS, Tuberculosis and Malaria. Der Fonds sammelt Gelder und kanalisiert sie in Gesundheitsprojekte weltweit. Doch auch hier ist das Budget knapp: Norbert Hauser, bis 2017 Vorsitzender des Verwaltungsrats des Globalen Fonds, betonte, dass für die nächste Wiederauffüllung (2020-2022) bislang 19 Milliarden Dollar fehlten.
Heiko Warnken vom Bundesministerium für wirtschaftliche Zusammenarbeit hielt auch die inhaltliche Ausrichtung für wichtig. Für die Stärkung von Gesundheitssystemen gibt der Global Fund maximal 40% seines Budgets aus, das könne deutlich verbessert werden. Auch sei eine bessere Abstimmung mit anderen Geldgebern und mit den Regierungen von Empfänger-Ländern notwendig. Nicht zu unterschätzen sei zudem das Problem der konkurrierenden Strukturen: China bereite derzeit einen neuen Afrika-Asien-Fonds vor – hier seien Absprachen wichtig, um unnötige Doppelstrukturen zu vermeiden.
Problematisch sei außerdem, dass bereitstehende Gelder oft nicht abgerufen würden. Länder wie Kenia würden nur einen Bruchteil der Global Fund Maßnahmen in Anspruch nehmen, weil die Verwaltung nicht funktioniere.
Diskutiert wurde die Frage, wie mit solchen Ländern umzugehen sei, die wirtschaftlich erstarkt sind und deshalb aus der Förderung des Global Fund herausfallen (sogenannte Transition Countries). Der Ausstieg aus dem Global Fund müsse besser vorbereitet werden. Die Nehmerländer müssten allerdings auch in die Verantwortung genommen werden, um die dauerhafte Versorgung ihrer Bevölkerung sicherzustellen.
Frank Mischo von der Kindernothilfe erinnerte daran, dass sich 2001 die afrikanischen Staaten verpflichtet haben, 15% des Staatshaushalts für Gesundheit aufzuwenden (Abuja-Ziel). Das Ziel ist bisher noch nicht erreicht, aber Malawi hat zum Beispiel in 15 Jahren die Gesundheitsausgaben vervierfacht, auf jetzt 11,4%.
Selbst ein deutlich reicheres Land wie Indien erreicht bisher nur 4,7%. Viele Gruppen profitieren aber nicht von dem staatlichen Gesundheitsetat – und das betreffe nicht nur Randgruppen, sondern besonders auch Frauen und Kinder.
Rolle der Zivilgesellschaft
Das 90-90-90-Ziel zu erreichen, ist demnach mit komplexen Herausforderungen verbunden. Die Zivilgesellschaft kann und muss hier einen wesentlichen Beitrag leisten. Zum einen, indem sie vulnerablen Gruppen eine Stimme gibt, auf deren Bedürfnisse hinweist und sich für grundlegende Werte wie Würde und Menschenrechte einsetzt. Zum anderen kann sie dazu beitragen die politischen Rahmenbedingungen zu verbessern – Bildung und Armutsbekämpfung als Grundlage für ein selbstbestimmtes Leben zu erreichen. In der Jubiläumskonferenz fand die frühere Entwicklungsministerin Heidemarie Wieczorek-Zeul dann auch deutliche Worte: Auch wenn viel erreicht worden sei – es sei bedenklich, dass die Social Development Goals offenbar für viele PolitikerInnen nach wie vor ein Tabu seien. (CW)
Artikel aus dem Pharma-Brief 1/2018, S. 3
Bild © Klaus Koch
[1] UNAIDS (2014) 90-90-90. An ambitious treatment target to help end the AIDS epidemic. www.unaids.org/sites/default/files/media_asset/90-90-90_en.pdf
[2] Pharma-Brief (2014) Wir haben noch lange nicht alle Probleme gelöst. Nr. 6, S. 3
[3] Deutsche Aidshilfe (o.J.) Positive Stimmen. Erlebnisbericht des PLHIV Stigma Index in Deutschland.
www.aidshilfe.de/shop/pdf/2482
[4] www.unaids.org/en/resources/909090
Bundesregierung hört zu
Beteiligung an globaler Gesundheitsstrategie
Bis Ende 2019 will die Bundesregierung eine Strategie zu globaler Gesundheit entwickeln. Der Prozess dahin ist von bislang ungewohnter Beteiligung und Transparenz geprägt. Auf einer Austauschveranstaltung am 5.9.2018 konnten die nichtstaatlichen Akteure ihre Positionen vorstellen.
Bereits in ihrer Koalitionsvereinbarung hatten die Regierungsparteien betont, dass Deutschland eine aktive und konstruktive Rolle in der internationalen Gesundheitspolitik spielen solle. Und auch Bundeskanzlerin Angela Merkel hat sich das Entwicklungsziel Gesundheit (Ziel 3 der nachhaltigen Entwicklungsziele der Vereinten Nationen) auf die Fahne geschrieben. Gemeinsam mit dem Präsidenten Ghanas, Nana Addo Dankwa Akufo-Addo, und der norwegischen Ministerpräsidentin Erna Solberg schrieb sie im April an Tedros Adhanom Ghebreyesus, den Generaldirektor der Weltgesundheitsorganisation (WHO).[1] Die drei StaatschefInnen mahnten in ihrem Brief verstärkte Anstrengungen für das Entwicklungsziel Gesundheit. Sie forderten die WHO auf, bei der Umsetzung eine Führungsrolle zu übernehmen und gemeinsam mit anderen UN-Organisationen einen konkreten Aktionsplan zu entwickeln.
Konsultationsprozess
Auch das Gesundheitsministerium (BMG) hat einen konstruktiven Dialog zum Thema globale Gesundheit in Gang gesetzt. Bereits Ende 2017 gab es eine erste Austauschveranstaltung zu globaler Gesundheit, die allerdings stark auf die G 20 Aktivitäten konzentriert war und wenig Raum für Debatten ließ. Auf einer weiteren BMG-Veranstaltung im Juni 2018 wurde dann vereinbart, dass die fünf verschiedenen Akteursgruppen (Jugend, Think Tanks, Wirtschaft, Wissenschaft und Zivilgesellschaft) eigene Positionspapiere erarbeiten.[2] Diese wurden am 5. September in Berlin vorgestellt und diskutiert. Das Memento Bündnis[3] und die Deutsche Plattform für globale Gesundheit (DPGG) [4] hatten weitere Papiere vorgelegt; in beiden Netzwerken engagiert sich auch die BUKO Pharma-Kampagne.
Kritischer Konsens
Erstaunlich waren die großen Übereinstimmungen zwischen den Papieren, obwohl sie unabhängig voneinander entwickelt worden waren. Das Prinzip der Universal Health Coverage (UHC), also eines umfassenden Zugangs zur Gesundheitsversorgung und zu sozialer Absicherung im Krankheitsfall war überall ein zentraler Punkt. Das Menschenrecht auf Gesundheit als Leitprinzip fand sich in allen Papieren – mit Ausnahme des Dokuments das von den privatwirtschaftlichen Akteuren vorgelegt wurde.
Viele Bereiche über den eigentlichen Gesundheitssektor hinaus sind für die Gesundheit wichtig und müssen mit bedacht werden, wenn es um die Vermeidung von Krankheiten und eine bessere Versorgung geht. Deshalb ist die Forderung nach „health in all policies“ nicht verwunderlich.
Ein wesentlicher Fortschritt ist, dass die nachhaltigen Entwicklungsziele (SDGs) im Gegensatz zu den vorherigen Millennium Development Goals (MDGs) für reiche und arme Länder gleichermaßen gelten. Eingefordert wurde deshalb, dass Deutschland sich nicht nur international konstruktiv für die Umsetzung der SDGs stark macht, sondern auch selbst seine Hausaufgaben macht. Angefangen davon, dass die Versorgung im Krankheitsfall für Geflüchtete und andere vulnerable Gruppen miserabel ist, über ungleiche Bildungschancen, Armut bis hin zum sozialen Wohnungsbau gibt es auch in Deutschland viele gesundheitsbezogene Baustellen.
Debatten
In der Diskussion wurde deutlich: Während intersektorale Ansätze befürwortet wurden, ist es bis zur praktischen Umsetzung noch ein weiter Weg. So muss zum Beispiel Klimaschutz und die Bekämpfung der Luftverschmutzung gegen die Interessen der Autoindustrie und Energiewirtschaft durchgesetzt werden. Zwar waren bei der Diskussion am 5.9.2018 nicht nur VertreterInnen des Gesundheits- und Entwicklungshilfeministeriums anwesend, sondern auch die Ressorts Forschung und Landwirtschaft sowie das Bundeskanzleramt. Aber weder das Ministerium für Wirtschaft noch das für Arbeit und Soziales waren vertreten.
Aber auch im klassischen Gesundheitssektor gibt es reichlich Reibungsflächen: Die Priorisierung von Patentschutz versus Zugang zu Arzneimitteln oder hohe Preise, die auch in Industrieländern die Versorgung gefährden. Die fehlende Forschung zu vernachlässigten Krankheiten erfordert andere Finanzierungsmodelle. All das wird sich nur gegen den Widerstand von Big Pharma erreichen lassen.
Die Zersplitterung der globalen Gesundheitspolitik war ebenfalls ein wichtiges Thema. Die ständig wachsende Zahl separater Initiativen wie dem „Globalen Fonds zur Bekämpfung von Aids, Tuberkulose und Malaria“, der Impfinitiative GAVI und vielen anderen führt zu vertikalen Interventionen, die viele Ressourcen fressen. Auch wenn diese Programme im Einzelfall deutliche Erfolge erzielen, besteht immer die Gefahr, dass die breite Versorgung der Bevölkerung, die ja nicht nur an diesen wenigen Krankheiten leidet, auf der Strecke bleibt. Es bleibt eine Herausforderung, solche vertikalen Programme in existierende Strukturen zu integrieren und die Stärkung der allgemeinen Versorgung als wichtige Teilaufgabe solcher Programme zu begreifen. Die Förderung von Universal Health Coverage muss nach Meinung der meisten TeilnehmerInnen Vorrang haben.
Rolle der Wirtschaft
Für die Industrie äußerte sich das „German Healtcare Partnership“ (GHP). Die Wirtschaft sieht UHC auch als lohnenden Markt. So bietet sie an, Gesundheitsversorgungseinrichtungen zu beraten oder sogar in eigener Regie zu übernehmen. Ihr Beitrag zur Entwicklung von Versicherungssystemen ist die Einführung von privaten Krankenversicherungen. Die „Lieferung der technischen (fachlichen) Expertise bei der Definition eines Katalogs zur medizinischen Grundversorgung […]“ lässt eher die Durchsetzung von Partikularinteressen erwarten als eine am Allgemeinwohl orientierte Auswahl von medizinischen Leistungen – vom Paternalismus eines solchen Ansatzes einmal ganz zu schweigen. Auch auf die im Papier versprochenen „innovative[n] Medikamente für die Bedürfnisse der Entwicklungsländer“ warten diese schon lange vergeblich.
Dass sich einige Firmen nach jahrelangem Druck von NGOs in bescheidenem Umfang auch für vernachlässigte Krankheiten engagieren, kann nicht darüber hinwegtäuschen, dass das Geschäftsmodell von Big Pharma die gesundheitlichen Bedürfnisse von großen Teilen der Weltbevölkerung nicht befriedigen kann. Insofern wirken die folgenden Sätze aus dem Wirtschaftspapier wie der blanke Hohn: „Innovative Gesundheitsprodukte sind Lösung und nicht Last für nachhaltige Gesundheitssysteme. Es gilt, Innovationen durch Stärkung bzw. Aufrechterhaltung geeigneter Anreizsysteme, insbesondere dem Schutz des geistigen Eigentums, zu fördern.“ Dabei hilft ja gerade der Patentschutz nicht, Medikamente für die Armen zu entwickeln, im Gegenteil fördert er die Entwicklung von Medikamenten für zahlungskräftige Märkte. Er sorgt außerdem dafür, dass hohe Arzneimittel durchgesetzt werden können, die mit den Herstellungskosten nichts zu tun haben.
„Public Private Partnership“ stehen aus Sicht der Wirtschaft im Mittelpunkt. Das hat ja auch einige Vorteile: Man muss selbst nicht so viel investieren, denn das meiste Geld stammt in der Regel aus öffentlichen Töpfen und man kann mitreden, wie die Prioritäten gesetzt werden. Dazu liefert die Wirtschaft auch ein Beispiel: „Ein besonders wirkungsvolles und erfolgreiches Beispiel für akteursübergreifende Zusammenarbeit ist der World Health Summit (WHS), er steht für die gute und enge Zusammenarbeit der deutschen Gesundheitswirtschaft mit der Wissenschaft.“ Dieses seit einigen Jahren in Berlin stattfindende Treffen wird von der M8 Alliance, einem Zusammenschluss von akademischen Zentren, in enger Zusammenarbeit mit der Wirtschaft durchgeführt. Es erhebt den Anspruch „die Gesundheit auf dem ganzen Planeten zu verbessern“ und „die Agenda von Morgen zu steuern, um Forschung, Ausbildung, Gesundheitsversorgung und policy outcomes zu verbessern“.[5] Die M8 Alliance ist ein exklusiver Club und repräsentiert vorwiegend WissenschaftlerInnen aus Industrieländern. Auf der Liste der RednerInnen sind auch in diesem Jahr Firmen und Public Private Partnerships gut vertreten.[6] Kein Wunder also, wenn einige Veranstaltungen eine kommerzielle Schieflage bekommen.
Immer wenn sich kommerzielle Interessen mit öffentlichen mischen, besteht die Gefahr der Vereinnahmung und Abschwächung von an sich sinnvollen Zielen. Im Englischen wurde dafür der Begriff „Engineering of Consent“ geprägt. Das Papier der Wirtschaft bietet dafür unfreiwillig eine Bestätigung: „Schulterschluss aller in Gesundheitsthemen involvierten Akteure (Wirtschaft, Wissenschaft, NROs und Politik): proaktive Abstimmung der Aktivitäten sowie gegenseitiger Erfahrungs- und Wissenstransfer können ein effizientes und wirtschaftliches Engagement aller Akteure unterstützen […]“.
Mit der Forderung nach einem „Schulterschluss“ standen die Industrievertreter auf der Austauschveranstaltung am 5.9. allerdings allein auf weiter Flur. Von verschiedenen TeilnehmerInnen – auch aus den Ministerien – wurde der Dissens als treibende Kraft in einer produktiven Debatte hervorgehoben.
Irritationen
Für einigen Unmut sorgte das Phantom eines „Global Health Hub Germany“ (GHH). Von den fünf am 5.9. anwesenden Akteursgruppen war offensichtlich lediglich die Wirtschaft an den Planungen für ein ständiges Austauschforum des BMG zu internationaler Gesundheitspolitik beteiligt und gut informiert. Es gab vorab keine offiziellen Informationen von Seiten des BMG. Lediglich einigen NGOs war ein paar Tage vor dem Treffen ein kurzes Konzeptpapier zugespielt worden, das eine vage Idee von dem Vorhaben gab. An das BMG sei „wiederholt der Wunsch einer Vernetzungsplattform für Akteure in der globalen Gesundheit herangetragen“ worden.[7] Es fehle „ein sektorübergreifender Austausch“. Der GHH solle „als zentrale Anlaufstelle in Deutschland für nationale und internationale Akteure im Bereich Globale Gesundheit dienen.“ Er „soll ein von der Bundesregierung unabhängiges selbständiges Forum sein […]. Ob und in welcher Form die Ressorts [außer dem BMG] sich einbringen wollen, ist diesen freigestellt.“ Auch die bisherigen Gesprächspartner zum GHH werden genannt: „WHS/Charité, GHP, Bill und Melinda Gates Stiftung, Wellcome Trust, VENRO etc.“. Dummerweise wusste aber Letzterer, ein Zusammenschluss von NGOs, gar nichts davon. Der Verband hatte erst durch das durchgesickerte Papier von seiner angeblichen Beteiligung am Global Health Hub erfahren. Die harsche Kritik an dieser Vorgehensweise veranlasste das Ministerium immerhin dazu, offiziell um Austausch zu bitten. Nach den Vorstellungen des BMG soll der GHH im Dezember 2018 vorgestellt werden.
Wie geht’s weiter?
Das BMG betonte, dass es die Zivilgesellschaft in die weitere Diskussion über die globale Strategie einbeziehen will. Dabei wurde als ein Format der GHH vorgeschlagen, aber über die Ausgestaltung könne noch geredet werden. Die Zivilgesellschaft wird sich gut überlegen müssen, ob sie einem Gremium Legitimität verleihen möchte, das nach bisherigem Planungsstand als Public Private Partnership angelegt ist, das politische Fortschritte in Richtung Global Health eher bremsen könnte. Die starke Industriepräsenz, die Rolle der Gates-Stiftung und die Ausrichtung auf Konsens sprechen dafür.
Weitere Diskussionen über die Global Health Strategie gab es schon wenige Tage später. Das BMG und das BMZ luden am 10.9. zu einem Dialogforum ein. Die Themen: „Konkret möchten wir uns mit Ihnen zu der Bekämpfung antimikrobieller Resistenzen im Sinne eines ‚One-Health‘-Ansatzes, der Gesundheitsförderung und Prävention nicht übertragbarer Krankheiten und den Potenzialen einer zunehmenden Digitalisierung des Gesundheitssektors austauschen.“ Allerdings war der Kreis der Eingeladenen mit 20 Organisationen deutlich kleiner als am 5.9. Und der Bundesverband der Deutschen Industrie war gleich zweimal eingeladen, einmal als BDI und einmal als German Healthcare Partnership. Auf der Teilnehmerliste fehlten VertreterInnen der Medizin und Public Health dagegen völlig. Dass dann doch noch ein Vertreter der Deutschen Plattform für Globale Gesundheit teilnehmen konnte, erwies sich als wichtig. In der Debatte wären sonst die Themen Antibiotikaresistenz (ABR) und nichtübertragbare Krankheiten (NCDs) völlig untergegangen. Dabei ist die Rate der vermeidbaren Todesfälle durch NCDs in Entwicklungs- und Schwellenländern sogar höher als in reichen Staaten, es handelt sich also um ein zentrales Thema für Global Health. Und auch bei Antibiotikaresistenzen sind entschiedene Maßnahmen in der Humanmedizin wie in der Tierhaltung essenziell. Die Lernkurve der Bundesregierung könnte also noch steiler werden.
Letztlich wird sich die globale Gesundheitsstrategie der Bundesregierung nicht nur daran messen, ob die Positionen auf dem Papier korrekt sind, sondern ob und wie sie umgesetzt werden. Bei der Austauschveranstaltung am 5.9. fand deshalb ein Vorschlag der Pharma-Kampagne im Publikum großen Beifall: Man solle doch alle künftigen politischen Vorhaben der Bundesregierung und vor allem neue Gesetzentwürfe einer Entwicklungsverträglichkeitsprüfung im Sinne der SDGs unterziehen. (JS)
Bild: Austauschveranstaltung mit dem BMG am 5. September in Berlin © Jörg Schaaber
Artikel aus dem Pharma-Brief 7/2018, S.1
[1] Merkel A et al. (2018) Letter to Dr. Tedros, 19 April www.bundesregierung.de/Content/DE/Artikel/2018/04/2018-04-19-merkel-solberg-akufo-addo.html
[2] www.bundesgesundheitsministerium.de/ministerium/meldungen/2018/september/globale-gesundheitspolitik.html#c13758
[3] Memento-Bündnis (2018) www.bukopharma.de/images/aktuelles/Memento-Buendnis_2018_Globale_Gesundheit.pdf
[4] DPGG (2018) www.bukopharma.de/images/aktuelles/dpgg_2018_deutsche_Strategie_globale_Gesundheit.pdf
[5] WHS (2018) Vision and goals. www.worldhealthsummit.org/about/vision-and-goals.html [Zugriff 11.9.2018]
[6] WHS (2018) Speakers. www.worldhealthsummit.org/conference/speakers.html [Zugriff 11.9.2018]
[7] BMG (2018) Projektskizze „Aufbau eines Global Health Hub Germany (GHH Germany)“
Buch: Riskante Manöver
Ein Pharmakonzern hat ein Problem. Sein neues Schmerzmittel für Kinder scheint schwere unerwünschte Wirkungen zu haben. Der PR-Agent Mats Holm wird engagiert, um den Schaden für die Firma zu begrenzen.
So steigt der WDR-Journalist Birand Bingül in seinen Erstlingskrimi „Riskante Manöver“ ein. Daraus entwickelt sich eine Geschichte, die zu jedem Krimi gehört: Verwicklungen, falsche Fährten, und etwas gewalttätig geht es natürlich auch zu.
Der Autor schafft es, mit seiner fiktiven Handlung einen glaubwürdigen Einblick in das Denken und Handeln der Pharmabosse zu geben. Ganz nebenbei bekommen die LeserInnen auch noch etliche Fakten und Zusammenhänge über die dunklen Seiten der Branche serviert, die – leider – den Tatsachen entsprechen. Auch wenn der Autor zu Recht betont, „alle in diesem Roman geschilderten Handlungen und Personen sind frei erfunden“, so ähnlich könnte eine Krise in der Pharma-Industrie ablaufen. Vermutlich etwas unblutiger, aber die Abwehrstrategien sind aus real existierenden Fällen durchaus bekannt. Schließlich hat der Autor bei seinen Recherchen für den Roman auch mit der BUKO Pharma-Kampagne und anderen Kennern der Szene gesprochen. (JS)
Artikel aus dem Pharma-Brief 3/2018, S. 7
Bild: Cover von Bingül B. (2018) Riskante Manöver. Ein Fall für Mats Holm. München: btb. 446 S., 10 €
Brustkrebs: Leere Versprechen
Palbociclib verlängert das Leben nicht
Palbociclib wurde im November 2016 gegen bestimmte Formen von Brustkrebs zugelassen. Obwohl zu diesem Zeitpunkt keine der drei Studien zum Wirkstoff abgeschlossen war, schürte der Hersteller Pfizer große Hoffnungen. Der Gemeinsame Bundesausschuss (G-BA) urteilte dagegen letztes Jahr: „kein Zusatznutzen“ (wir berichteten [1] ). Jetzt wurden die Endergebnisse einer weiteren Studie [2] bekannt. Auch sie konnte nicht zeigen, dass Frauen durch Palbociclib länger leben.
Noch im Februar 2018 kritisierte der Berufsverband der niedergelassenen gynäkologischen Onkologen (BNGO) die Entscheidung des G-BA, Palbociclib keinen Zusatznutzen zu bescheinigen. Das Präparat habe „eine ungefähre Verdopplung der Zeit bis zum Fortschreiten der Erkrankung gezeigt.“ [3] Doch tatsächlich waren zur Ermittlung des „progressionsfreien Überlebens“ nur Röntgenbilder zum Tumorwachstum ausgewertet worden. Die Studien konnten keine Verbesserung bei den Krankheitssymptomen belegen.
Der Verband plädierte dafür, bei der Bewertung auch die Lebensqualität zu berücksichtigen. Das hatte der G-BA bei seiner Entscheidung 2017 durchaus getan: Ein Vorteil für Palbociclib ließ sich so nicht erkennen. Dafür ergab die Auswertung der unerwünschten Wirkungen einen deutlichen Nachteil für das neue Medikament.[4]
Am 25. Juni 2018 veröffentlichte der Hersteller von Palbociclib eine Pressemitteilung, die sich allerdings lediglich an Investoren richtete: „Pfizer verkündet Ergebnisse zum Gesamtüberleben bei der Phase 3 Studie PALOMA-3“. [5] Wie schon die Überschrift ahnen lässt, waren die Ergebnisse nicht wie erhofft ausgefallen. Etwas verschämt wird im Text mitgeteilt, dass kein signifikanter Überlebensvorteil gezeigt werden konnte. Pfizer versucht das allerdings mit der Aussage schönzureden, dass es „einen positiven Trend“ gebe.
Shareholder value
Dass statt der medizinischen Fachwelt zuerst die Investoren informiert werden, hat einen schlichten Grund: Warnt eine Firma nicht rechtzeitig, dass ein Produkt die wirtschaftlichen Erwartungen nicht erfüllt, können Aktionäre in den USA auf Schadenersatz klagen. Über einen ähnlichen Fall bei einem anderen Wirkstoff, der inzwischen wieder vom Markt genommen wurde, hatten wir berichtet.[6]
Die Bewertung des G-BA von 2017 ist teilweise befristet, weil die endgültigen Ergebnisse von zwei Studien mit verschiedenen Patientinnengruppen ausstehen.[7] Die erste Frist endet am 1.10.2018 und bezieht sich auf die Studie Paloma 3, deren Ergebnisse nun bekanntgegeben wurden. Die Ergebnisse für die dritte und letzte Studie (Paloma 2) werden Ende 2018 erwartet. Deren vom G-BA ausgewertete Zwischenergebnisse ergaben keinen Überlebensvorteil.[4] Endgültige Daten muss der Hersteller bis zum 1.3.2019 vorlegen.
Anderthalb Jahre nach der Zulassung
hat also noch keine Studie nachgewiesen, dass Frauen mit Brustkrebs dank Palbociclib länger leben. Die Nebenwirkungen sind erheblich. Das ist insofern besonders relevant, weil der neue Wirkstoff zusätzlich zur bisher üblichen
Therapie gegeben wird. Nachdem nun die Endergebnisse für zwei Studien vorliegen, erscheint die Nutzen-Schaden Bilanz für mindestens einen Teil der Patientinnen negativ.[8] Da stellt sich immer mehr die Frage, warum die europäische Arzneimittelbehörde EMA Palbociclib zu einem Zeitpunkt zugelassen hatte, zu dem es noch keinerlei Daten zum Überleben gab.
Verfrühte Zulassung
Basis der Zulassung war das progressionsfreie Überleben (PFS), also ein verlangsamtes Wachstum von Tumoren. Dies ist nur ein Surrogat (Ersatz), das Hinweise auf relevante Ergebnisse wie längeres Überleben oder Linderung der Krankheitssymptome erlauben soll. Das PFS ist insgesamt leichter und schneller zu erheben als das tatsächliche Überleben, das eine viel längere Beobachtungsdauer erfordert.
Das Surrogat progressionsfreies Überleben hat sich damit zum wiederholten Male als untauglicher Indikator für einen relevanten Nutzen herausgestellt. Das zeigte eine Übersichtarbeit aus den USA: Mehrere Jahre nach der Zulassung konnte nur für 14% der Krebsmedikamente, die auf Basis von Surrogaten zugelassen waren, ein Überlebensvorteil belegt werden. Bei der Hälfte der Medikamente stellte sich letztlich heraus, dass Patientinnen durch den neuen Wirkstoff nicht länger leben, bei den übrigen ist das nach wie vor unklar. [9],[10]
Shareholder first
Für Pfizer hatte die Information von Aktionären über die ungünstigen Ergebnisse offensichtlich Vorrang vor der Unterrichtung von Ärztinnen und Ärzten. Das ist ein Symptom dafür, dass Investoren mit einem besseren Schutz ihrer Interessen rechnen können als PatientInnen. Es wäre an der Zeit, dass die EMA ihre eigenen Zulassungsentscheidungen kritisch hinterfragt. (JS)
Artikel aus dem Pharma-Brief 6/2018, S. 4
Bild © Jörg Schaaber
[1] Pharma-Brief (2017) Viel Lärm um nichts? Nr. 4, S. 4
[2] Pfizer hat insgesamt drei zulassungsrelevante Studien durchgeführt, Paloma 1, 2 und 3.
[3] Deutsches Ärzteblatt (2018) Berufsverband kritisiert G-BA-Bewertung neuer Krebstherapeutika. News 18. Feb. www.aerzteblatt.de/treffer?mode=s&wo=17&typ=1&nid=89322&s=palbociclib
[4] G-BA (2017) Beschluss zu Palbociclib vom 18. Mai www.g-ba.de/informationen/nutzenbewertung/269/#tab/beschluesse
[5] Pfizer (2018) Pfizer Announces Overall Survival Results from Phase 3 PALOMA-3 Trial of IBRANCE® (Palbociclib) in HR+, HER2- Metastatic Breast Cancer. Investor news press release 25 June
[6] Pharma-Brief (2017) … und wieder die Aktionäre zuerst. Nr. 3, S. 6
[7] Für eine weitere Patientinnengruppe legte der Hersteller keine Daten vor. Hier gilt der Beschluss des G-BA „kein Zusatznutzen“ unbefristet.
[8] Für eine Patientinnengruppe hatte Pfizer dem G-BA keine Daten vorgelegt, für eine weitere Gruppe sind die Ergebnisse der noch nicht abgeschlossenen Paloma 2 Studie relevant.
[9] Pharma-Brief (2017) Bescheidener Fortschritt Nr. 8-9, S. 1
[10] Kim C und Prasad V (2015) Cancer Drugs Approved on the Basis of a Surrogate End Point and Subsequent Overall Survival .JAMA Int Med; 175, p 1992
Belohnt wird später
Interessenkonflikte bei US-Arzneimittelzulassung
Die US-FDA lädt regelmäßig ExpertInnen ein, die ihre Ansichten über anstehende Zulassungen äußern. Das US-Wissenschaftsmagazin Science deckte jetzt auf, dass es ein System der nachträglichen Belohnung gibt.[1] Nicht nur ExpertInnen profitieren davon, auch MitarbeiterInnen der FDA.
Auch wenn das Votum des Advisory Committee für die Behörde nicht bindend ist, folgt sie meist der Ansicht der Fachleute. Wer an einem solchen Treffen der FDA teilnimmt, muss eine Erklärung zu Interessenkonflikten ausfüllen. Wenn die FDA sie als relevant ansieht, werden sie bekanntgegeben.
Ist jedoch noch kein Geld geflossen, kann auch nichts offengelegt werden. Doch auffällig häufig erhalten ExpertInnen aus Advisory Committees später erhebliche Summen von Pharmafirmen. Der, dessen Stimme im Zulassungsprozess gehört wird, ist für Hersteller interessant. Das gilt nicht nur für die Firma, die von einer positiven Expertenmeinung im Zulassungsprozess profitiert hat, sondern auch für Firmen, die Konkurrenzprodukte für dieselbe Indikation entwickeln.
Science wertete über 20 Advisory Committees der FDA aus. Insgesamt hatten dabei 107 ÄrztInnen abgestimmt, die keine Interessenkonflikte angegeben hatten. 66 erhielten später Pharmagelder, 40 davon über 10.000 US$, sieben sogar mehr als eine Million US$.
Drehtür FDA – Industrie
Auch die MitarbeiterInnen der FDA, die die Zulassungsanträge inhaltlich bearbeiten, sogenannte Reviewers, laufen Gefahr, bei ihrer Arbeit künftige Jobs im Auge zu haben. Bereits 2016 hatten Jeffrey Bien und Vinay Prasad die spätere Karriere von allen Reviewern für Krebsmedikamente von 2006-2010 untersucht.[2] Die Hälfte war auch 2016 noch bei der FDA beschäftigt, aber von den Ausgeschiedenen hatten 57,7% inzwischen einen Job bei der Industrie oder fungierten als Berater für Pharmafirmen.[3]
Science wertete aktuelle Daten aus. Die Redakteure deckten auf, dass von 16 Reviewers, die die FDA verließen, 11 entweder direkt bei einer Pharmafirma anfingen oder als Berater für die Industrie tätig wurden.[4] Einziger Schutzwall gegen den Drehtüreffekt: Leitende MitarbeiterInnen der Behörde dürfen nach dem Wechsel in die Industrie ein bis zwei Jahre die Firma nicht bei der FDA vertreten. Das gilt aber nicht für gewöhnliche Reviewer, für die es keine Einschränkungen in der weiteren Berufsausübung gibt.
Mit einigen Beispielen illustriert Science die Problematik von scheinbar unabhängigen ExpertInnen, die sich später von Firmen unterstützen lassen und von BehördenmitarbeiterInnen, die die Seiten wechseln.
Ticagrelor
Vier Ärzte, die 2010 beim FDA Advisory Committee über den Gerinnungshemmer Ticagrelor abstimmten (Markenname in den USA: Brilinta®), hatten zu diesem Zeitpunkt laut FDA keine relevanten Interessenkonflikte. Aber in den folgenden Jahren ergoss sich über die Vier eine Gelddusche von AstraZeneca und Konkurrenten. Besonders viel bekam der Kardiologe Jonathan Halperin ab: Von 2013-2016 erhielt er über 200.000 US$ als Honorare, für Reisekosten und Beratung.[5] Für Forschung zu Ticagrelor, an der Halperin persönlich beteiligt war, zahlte AstraZeneca seiner Uni fast zwei Millionen US$. Der Kardiologe sieht für sich persönlich kein Problem. Wenn eine Firma ihn für einen Vortrag oder Beratung bezahle, „ist das wirklich nicht viel anders als wenn dir die Versicherung einen Scheck dafür gibt, dass du irgendwann einen Patienten behandelt hast.“ Halperin räumte immerhin ein, dass die Erwartung auf zukünftige Belohnung Ansichten beeinflussen kann: „Ich teile die Sorge, dass das dazu führen kann, dass Leute auf eine Weise agieren, die man nicht möchte.“
Quetiapin
2009 gab es gleich zwei FDA Advisory Committees zu Quetiapin (Seroquel® von AstraZeneca). Dabei ging es um die Frage, ob Quetiapin künftig auch gegen Schizophrenie und bipolare Störungen eingesetzt werden dürfe. Bereits damals war bekannt, dass der Wirkstoff, wenn er mit anderen Medikamenten kombiniert wird, plötzlichen Herzstillstand auslösen kann. Trotzdem stimmten die beratenden ÄrztInnen mit großer Mehrheit beiden Zulassungserweiterungen zu. Mehrere der Berater erhielten anschließend erhebliche Summen von der Industrie.
Mit 1,36 Mio. US$ kassierte Christopher Granger den größten Betrag. Er behauptete auf Nachfrage, das Geld sei nur in die Forschung geflossen. Allerdings sagt die staatliche Datenbank etwas anderes: Über 400.000 US$ flossen als Honorare, für Reiskosten und für Beratung an Granger persönlich – darunter das ganze Geld, das er von AstraZeneca erhalten hatte.
Granger rechtfertigte sich. „Ich bin mir darüber im Klaren, dass, wenn mich jemand bezahlt, mich das – wie jedes andere menschliche Wesen – in meiner Denkweise beeinflussen kann. Ich bin nicht so naiv.“[1] Trotzdem habe er geglaubt, dass für einige PatientInnen mit schweren psychischen Störungen der Nutzen von Quetiapin gegenüber den Risiken überwiege.
Auch ein FDA-Mitarbeiter fiel bei einem der Advisory Committee Meetings zu Quetiapin 2009 auf: Thomas Laughren, seinerzeit Direktor der Abteilung Psychopharmaka bei der FDA, kanzelte den Wissenschaftler Wayne Ray, der seine Untersuchung zum plötzlichen Herzstillstand vorgestellt hatte, regelrecht ab. Laughren hielt dagegen die Auswertung der Studien von AstraZeneca für glaubwürdig, in denen kein Risiko erkennbar war. Ray warnte jedoch davor, diese Aussage der Firma als „endgültig“ anzusehen. Methodisch seien die Berechnungen unzuverlässig, da AstraZeneca die Daten aus unterschiedlichen Studien einfach zusammengerechnet hatte, als sei es eine einzige Studie gewesen. Laughren entgegnete flapsig, plötzlicher Tod wäre „ein ziemlich endgültiges Ereignis“.
Kurz nach den Anhörungen verließ Laughren die FDA und gründete eine Beratungsfirma., die auch AstraZeneca bei Zulassungen half. Science wollte er keine Auskunft über seinen Rollenwechsel geben.
2010, also ein Jahr nach der Zulassungserweiterung, musste AstraZeneca dem Staat 520 Mio. US$ wegen Unregelmäßigkeiten bei klinischen Studien und wegen der Bewerbung von Seroquel® für nicht zugelassene Indikationen zahlen. Im gleichen Jahr erzielte die Firma – die trotz der großen Zahlung jedes Fehlverhalten abstritt – mit dem Medikament fünf Milliarden US$ Umsatz. Ein Jahr später, 2011, musste sie auf Anordnung der FDA eine Warnung in den Beipackzettel aufnehmen, dass bei Kombination mit anderen Medikamenten die Gefahr von Herzstillstand besteht.
Karen Birmingham, Pressesprecherin von AstraZeneca, sieht die Rolle von ehemaligen Behördenmitarbeitern dennoch positiv. Sie „bringen die Perspektive von erfahrenen Regulierern ein“ und würden damit den heutigen MitarbeiterInnen der Zulassungsbehörde helfen, „herausfordernde Entscheidungen über die Zulassung innovativer Arzneimittel, die Behandlungslücken schließen, zu treffen.“ [4]
Vinay Prasad, der die erste Untersuchung zum Drehtüreffekt bei der FDA durchgeführt hat, sieht das etwas anders. Schwache Regeln zu Interessenkonflikten bei der FDA und künftige Beschäftigungsaussichten, brächten die Bewertungen der Behörde in eine Schieflage. „Wenn dein möglicher nächster Arbeitgeber Nr. 1 dir gegenübersitzt, dann gibst du nicht den harten Hund, wenn du ihn reglementierst. Das liegt einfach in der menschlichen Natur.“ [4]
Laxe Kontrolle
Science überprüfte aber nicht nur spätere Zahlungen an ÄrztInnen, sondern auch, ob die Angaben zu Interessenkonflikten bei der FDA angemessen waren. Hier wurde ebenfalls mangelnde Kontrolle deutlich.
Bei vielen ExpertInnen, bei denen die FDA keine relevanten Konflikte sah, gab es in Wirklichkeit doch welche. Durch die Recherche wurde aufgedeckt, dass doch Geld floss: Firmen, die von der Zulassungsentscheidung betroffen waren, hatten die scheinbar unabhängigen ExpertInnen unterstützt.
Von den 17 durch die FDA als unabhängig deklarierten ÄrztInnen, die nach den FDA-Beratungen die höchsten Beträge von Firmen kassierten (über 300.000 US$) hatten 11 in Wirklichkeit auch schon im Jahr vor oder während der FDA-Beratung Gelder erhalten. Fünf davon von genau der Firma, um deren Produkt es bei der FDA ging. Science hatte diese Information in den Erklärungen zu Interessenkonflikten bei Fachartikeln der ÄrztInnen gefunden.
Ob das Versagen bei der FDA oder bei den beteiligten Ärzten liegt, bleibt unklar. Die beiden oben erwähnten Experten Halperin und Granger hatten zunächst zugesagt, Science ihre bei der FDA eingereichten Erklärungen zu Interessenkonflikten zur Verfügung zu stellen. Aber auch auf mehrfache Nachfragen wurden sie nicht zugesandt. Bei der FDA selbst wurde Science ebenso wenig fündig. Beide Erklärungen seien nicht auffindbar, teilte die Behörde mit.
Und in Europa?
Bei schwierigen Entscheidungen greift auch die europäische Zulassungsbehörde EMA auf externe ExpertInnen zu. Allerdings ist – im Gegensatz zu den USA – nicht ohne weiteres nachvollziehbar, wann das geschieht und wer angehört wurde. Deshalb helfen die auf der EMA Website hinterlegten Erklärungen zu Interessenkonflikten nicht weiter. Außerdem ist eine externe Überprüfung der Pharmazahlungen in Europa nicht möglich. In den USA konnte Science eine solche Untersuchung durchführen, weil durch den Physicians Payment Sunshine Act alle Zahlungen der Industrie an ÄrztInnen in einer öffentlichen Datenbank hinterlegt sind. Eine freiwillige Offenlegung, wie die seit kurzem in Deutschland eingeführte, erfasst nur einen kleinen Teil der tatsächlichen Zahlungen und ist deshalb nutzlos. (JS)
Artikel aus dem Pharma-Brief 6/2018, S.1
[1] Piller C and You J (2018) Hidden conflicts? Pharma payments to FDA advisers after drug approvals spark ethical concerns. Science. www.sciencemag.org/news/2018/07/hidden-conflicts-pharma-payments-fda-advisers-after-drug-approvals-spark-ethical [Zugriff 18.7.2018]
[2] Bien J und Prasad V (2016) Future jobs of FDA’s haematology-oncology reviewers. BMJ; ae54, p i5055
[3] Einige wenige wechselten zu anderen Behörden. Bei 30,8% konnte der neue Arbeitgeber nicht identifiziert werden
[4] Piller C (2018) FDA’s revolving door: Companies often hire agency staffers who managed their successful drug reviews. Science http://www.sciencemag.org/news/2018/07/fda-s-revolving-door-companies-often-hire-agency-staffers-who-managed-their-successful [Zugriff 18.7.2018]
[5] Die US-Datenbank mit Zahlungen der Industrie an ÄrztInnen wurde erst 2013 eingeführt, deshalb liegen zu vorangegangenen Jahren keine Daten vor.
Arm und unversorgt
Internationaler Währungsfonds schadet Gesundheit
Der Internationale Währungsfonds (IWF) ist in der Vergangenheit immer wieder für seine Auflagen bei der Kreditvergabe für Entwicklungsländer kritisiert worden. Sie trafen vor allem die ärmsten Teile der Bevölkerung und verschlechterten die gesundheitliche Lage. Wird jetzt alles besser?
Im vergangenen Jahr versuchte der IWF sein negatives Image aufzupolieren. In den letzten Jahren habe die Kreditvergabe sich nicht negativ auf die Ausgaben für Gesundheit ausgewirkt, im Gegenteil sie seien oft sogar gestiegen, behaupteten Mitarbeiter und auch IWF-Chefin Christine Lagarde in offiziellen IWF-Blogs.[1],[2] Diese Aussage haben Gino Brunswijck und Jesse Griffiths vom European network on debt and development (eurodad) unter die Lupe genommen.[3]
Sechs Studien nennt der IWF als Beleg für seine These. Doch die meisten Quellen unterstützen die Behauptung des größten Kreditgebers für Staaten nicht. Der einzige Artikel, der zu positiven Ergebnissen gelangt, stammt von den Autoren des Blogs selbst und basiert auf einem Bericht des IWF. Eine weitere als angeblicher Beleg zitierte Studie[4] widerspricht den Behauptungen des Fonds diametral. Sie kommt zu dem Schluss, dass „der IWF-Bericht methodische Fehler enthält, übermäßig optimistisch und potenziell irreführend ist.“ Die Wissenschaftler aus Cambridge werteten die Zahlen des IWF neu aus. Ihr Fazit ist: „Jedes zusätzliche Jahr, in dem ein Land an einem [Kredit-] Programm des IWF teilnimmt, bedeutet eine Senkung des Anteils der Gesundheitsausgaben am Bruttosozialprodukt um 1,7%.“
Dieses Jahr will der IWF seine Kriterien für die Kreditvergabe überarbeiten. Bei seiner verzerrten Sicht ist es allerdings fraglich, ob er dem Nachhaltigen Entwicklungsziel 3 „Gesundes Leben und Wohlergehen für Alle in jedem Alter“ gerecht wird. eurodad fordert aber noch tiefgreifendere Änderungen: „Die Auflagen des IWF sind oft hochkontrovers und stellen einen tiefen Eingriff in Schlüsselfragen der Wirtschaftspolitik dar. Die sollten eigentlich der Kern einer demokratischen Debatte des Landes sein und nicht von Washington vorgeschrieben werden.“[3] (JS)
Artikel aus dem Pharma-Brief 6/2018, S. 3
[1] Gupta S and Shang B (2017) Public spending on health care under IMF-supported programs. IMF blog https://blogs.imf.org/2017/03/09/public-spending-on-health-care-under-imf-supported-programs/
[2] Lagarde C (2017) Protecting Education and Health Spending in Low-Income Countries. IMF blog https://blogs.imf.org/2017/06/06/protecting-education-and-health-spending-in-low-income-countries/
[3] Griffith J and Brunswijck G (2018) IMF conditionality: still undermining healthcare & social protection? www.eurodad.org/IMF-conditionality-undermining-healthcare
[4] Stubbs T and Alexander Kentikelenis A (2017) Targeted social safeguards in the age of universal social protection: the IMF and health systems of low-income countries. Critical Public Health. DOI: 10.1080/09581596.2017.1340589
Antibiotika-Forschung koordiniert
Bundesregierung finanziert globale Zentrale
In Berlin entsteht derzeit ein Sekretariat, das weltweit die Entwicklung neuer Antibiotika koordinieren soll. Die Bundesregierung übernimmt für die ersten drei Jahre die Finanzierung. Damit löst sie eine Zusage vom G20-Gipfel in Hamburg 2017 ein. Dort wurde beschlossen, wegen fehlenden kommerziellen Interesses verstärkt öffentlich finanzierte Aktivitäten zu fördern.[1]
Die neue Einrichtung wird keine eigene Forschung und Entwicklung betreiben, sondern bestehende weltweite Aktivitäten miteinander vernetzten. Dazu sollen Regierungen ihre Förderprogramme aufeinander abstimmen und mit anderen Geldgebern, vor allem philanthropischen Stiftungen, zusammenarbeiten. Das Sekretariat mit dem sperrigen Namen „Global Antimicrobial Resistance R&D Hub“ wird mit 4 bis 5 Personen besetzt und am Deutschen Zentrum für Infektionsforschung in Berlin angesiedelt.[2]
Gründungsmitglieder sind unter anderem Russland, China, USA und Frankreich, die Europäische Kommission, die Gates Foundation und der Wellcome Trust.[3] Relevante überstaatliche Organisationen wie die Weltgesundheitsorganisation WHO oder die Ernährungs- und Landwirtschaftsorganisation der Vereinten Nationen FAO haben Beobachterstatus und sollen an den Board-Sitzungen teilnehmen.
An die Arbeit
Übergeordnetes Ziel ist, die Pipeline mit neuen Wirkstoffkandidaten zu füllen, um auch in Zukunft resistente Erreger behandeln zu können. Das soll laut Arbeitsplan mit mehreren Aktivitäten gefördert werden.[4] Eine öffentlich zugängliche Datenbank wird geschaffen, die alle relevanten Förderprogramme weltweit möglichst in Echtzeit abbilden soll. Auf dieser Basis sollen Prioritäten für Forschung und Entwicklung gesetzt und die Programme besser aufeinander abgestimmt werden. Leitprinzipien sind der „One Health“ Ansatz sowie die Verfügbarkeit der neuen Medikamente und Diagnostika für möglichst viele Menschen. Die Forschungsförderung soll mit einer Kombination von Push- und Pull-Mechanismen arbeiten.
Alle Mitglieder sind im Board vertreten und treffen sich regelmäßig. Die Mitglieder verpflichten sich, mit eigenen messbaren Aktivitäten für das Erreichen der Ziele zu arbeiten. So hat die deutsche Ministerin für Bildung und Forschung, Anja Karliczek, bei der offiziellen Gründung des AMR Hub im Mai 2018 angekündigt, die Bundesregierung werde in den kommenden zehn Jahren bis zu 500 Mio. Euro für die Forschung zur Resistenzproblematik bereitstellen.[5] (CW)
Artikel aus dem Pharma-Brief 6/2018, S. 6
[1] G20 (2017) Abschlusserklärung www.bundesgesundheitsministerium.de/fileadmin/Dateien/3_Downloads/E/Erklaerungen/G20-Abschlusserklaerung_der_Staats-_und_Regierungschefs.pdf
[2] Global AMR R&D Hub (2018) Terms of Reference www.gesundheitsforschung-bmbf.de/files/GLOBAL_AMR_RD_HUB_Terms_of_Reference.pdf
[3] BMBF (2018) A global effort to fight resistant pathogens. Pressemitteilung 22.5 www.bmbf.de/files/PM%200522-041%20Global_AMR_Hub_engl.pdf
[4] Global AMR R&D Hub (2018) Provisional Workplan 2018-2021 www.gesundheitsforschung-bmbf.de/files/Provisional_Work_Plan_2018-2021.pdf
[5] www.gesundheitsforschung-bmbf.de/en/GlobalAMRHub.php (Abruf 3.7.2018)
Alles auf Zucker
Neuer Online-Kurs der Pharma-Kampagne zu Diabetes im globalen Süden
Die Weltgesundheitsorganisation WHO listet Diabetes als eine der zehn häufigsten Todesursachen weltweit. Gerade ärmere Länder sind immer stärker betroffen. Doch die deutsche Entwicklungszusammenarbeit (EZ) wird dieser neuen Herausforderung kaum gerecht. Die BUKO Pharma-Kampagne hat einen E-Learning Kurs erstellt, der Abhilfe schaffen und EZ-MitarbeiterInnen für die Thematik sensibilisieren will. Pünktlich zum Weltdiabetestag am 14. November gingen die neuen Lernmaterialien online.
Diabetes als Wohlstandskrankheit des globalen Nordens – dieses Bild ist längst hinfällig. Globalisierung und Urbanisierung haben für steigende Diabetesraten rund um den Globus gesorgt. 1980 zählte die WHO 100 Millionen Zuckerkranke, heute sind es rund 422 Millionen. Mehr als 80% der Betroffenen leben mittlerweile in Ländern niedrigen und mittleren Einkommens. Eine BUKO-Umfrage unter deutschen Nichtregierungsorganisationen (NGO) zeigte auf, dass MitarbeiterInnen der medizinischen und humanitären Hilfe diese Veränderungen in ihren Projekten spüren. Gleichzeitig offenbarten sich jedoch deutliche Lücken in den Programmen der NGOs, etwa bei Diabetes in der Schwangerschaft oder dem Zusammenhang zwischen Diabetes und Tuberkulose.
Der neue Online-Kurs richtet sich daher an Personen, die im globalen Süden in Gesundheitsprojekten tätig sind oder in Deutschland zuständig für deren Konzeption und Management. Aber auch politische Akteure werden angesprochen, ebenso wie Projektförderer und Geldgeberorganisationen der Entwicklungszusammenarbeit. Das Lernmodul soll dazu beitragen, die Versorgung von PatientInnen, aber auch die Prävention im globalen Süden zu verbessern. Das neue Diabetes-Tool ist bereits der dritte Online-Kurs der BUKO Pharma-Kampagne. 2015 erschien unser E-Learning-Kurs zum Thema Verhütung, 2016 wurde ein Kurs zu Antibiotikaresistenzen veröffentlicht. Der neue Kurs ist frei zugänglich hier. Nach erfolgreicher Bearbeitung erhalten AbsolventInnen des Kurses ein Zertifikat.
Sechs Module vermitteln einen facettenreichen Überblick zur Erkrankung Diabetes. Jeder Abschnitt wird mit einem Erklärvideo eingeleitet. Das Tool führt dabei von medizinischem Grundwissen über die Diagnose bis hin zu einer rationalen Antidiabetika-Auswahl. Berichte aus dem globalen Süden – aus großen Staaten wie Nigeria, aber auch aus kleinen Ländern wie Tonga veranschaulichen lokale Besonderheiten, Ursachen und Folgen des Diabetes mellitus. Auch verschiedenartige politische Gegenmaßnahmen finden Erwähnung, etwa im Bereich Ernährung (Chile) oder Management von Komorbiditäten (Malawi). Praktische Hilfestellungen für die Zielgruppen runden den Kurs ab. TeilnehmerInnen können ein Zertifikat erhalten, wenn sie sich anmelden und Fragen zu den Modulinhalten beantworten.
Zur Konzeption und inhaltlichen Gestaltung des Tools veranstaltete die Pharma-Kampagne in Bielefeld drei ExpertInnentreffen mit Fachkräften aus Pharmazie, Entwicklungshilfe, medizinischer Nothilfe und Wissenschaft.
Der Weltdiabetestag bildete den passenden thematischen Rahmen für die Veröffentlichung des Lern-Werkzeugs. Seit 2006 wird der Aktionstag offiziell von den Vereinten Nationen begangen. Dabei markiert das Datum, der 14. November, den Geburtstag von Sir Frederick Banting. Er entdeckte 1922 zusammen mit Kollegen das Insulin. (MK)
Abgekartetes Spiel
Global Health Hub Germany ist keine gute Idee
Am Rande der Debatte um die globale Gesundheitsstrategie der Bundesregierung sickerten im September erste Informationen über ein neues Gremium durch: Der sogenannte Global Health Hub soll eine wichtige Rolle in der weiteren Entwicklung und Umsetzung der Gesundheitsstrategie bekommen (wir berichteten[1]). Je mehr Informationen über das geplante Diskussionsforum bekannt werden, umso fragwürdiger erscheint es.
Nach wie vor ist vage, was der „Global Health Hub Germany“ (GHHG) bezwecken soll. Ein Konzeptpapier vom 1. Oktober, das die staatliche Deutsche Gesellschaft für Internationale Zusammenarbeit (GIZ) im Auftrag des Gesundheitsministeriums verfasst hat, bleibt blumig: „Ein Global Health Hub Germany könnte als Dialog- und Vernetzungsplattform dienen und den Austausch und die Zusammenarbeit aller interessierten Akteure im Bereich Globale Gesundheit unterstützen.“ [2] „Erste Ideen zur Zielsetzung“ (siehe Kasten) sind ein bunter Strauß von Aktivitäten, die von Funktionen einer Denkfabrik über die Förderung von „Public Private Partnerships“ bis hin zur stromlinienförmigen Ausrichtung der globalen Public Health Debatte reichen.
Gremium mit Einfluss
Es wird zwar betont: „Der Global Health Hub Germany soll ein von der Bundesregierung unabhängiges Forum sein und als Anlaufstelle für Akteure der Globalen Gesundheit fungieren und deren sektorübergreifende Vernetzung ermöglichen“, aber das heißt noch lange nicht, dass der GHHG die Positionen der Bundesregierung nicht nachhaltig beeinflussen könnte.
Meinungsführerschaft
Einen Hinweis auf die Zielrichtung des GHHG liefert vor allem dessen Entstehungsgeschichte. Zwei Quellen haben unabhängig voneinander bestätigt, dass die Idee für den GHHG hauptsächlich von der Industrielobby ausging. Auch die Gates-Stiftung wurde schnell mit ins Boot geholt. Man könnte das Gremium als Versuch der Wirtschaft deuten, ihre Meinungsführerschaft in der globalen Gesundheitsdebatte nicht zu verlieren.
Pharma-, Agrar- und Nahrungsmittelindustrie stellen sich gern als Lösung für die Gesundheitsprobleme der Weltbevölkerung dar, obwohl ihre Aktivitäten durchaus auch gegenteilige Effekte haben. Gar nicht zu reden von direkt gesundheitsschädlichen Branchen wie fossilen Energiefirmen, der Chemie-, Auto- und Waffenindustrie. Bessere Gesundheit lässt sich aber nur erreichen, wenn negative Effekte wirtschaftlichen Handelns ebenso wie notwendige regulierende Maßnahmen offen debattiert werden. Von einem Diskussionsforum, in dem die Wirtschaft mit dem Bundesverband der deutschen Industrie und dem Verband forschender Arzneimittelhersteller[3] gleich doppelt vertreten ist, ist das kaum zu erwarten.
Fait accompli
Die Zusammensetzung des GHHG war von vorneherein festgelegt: Industrie, Gates und Welcome Stiftung, World Health Summit (eine firmengesponserte Veranstaltung in Trägerschaft von akademischen Institutionen, die vorwiegend in Industrieländern angesiedelt sind). Dazukommen sollten einige NGOs und wissenschaftliche Institutionen (die anfänglich von ihrem Glück gar nichts wussten, obwohl sie längst in den Strategiepapieren standen). Dieser Stakeholder-Ansatz verwischt wesentliche Unterschiede: NGOs orientieren sich am Gemeinwohl, die Wirtschaft ist an Gewinninteressen ausgerichtet. Auch die personellen und finanziellen Möglichkeiten der Stakeholder unterscheiden sich erheblich.
Im Oktober führte die GIZ eine Online-Befragung der Interessenträger zum GHHG durch. Der Fragenkatalog ließ durchweg nur positive Antworten zu. Die maximal mögliche Kritik wäre gewesen, dass man im GHHG keinen Mehrwert für sich selbst sieht (und dann nicht mitmacht). Zur Diskussion stand in keiner Weise, ob das Gremium an sich sinnvoll ist. Die Pharma-Kampagne und einige andere NGOs haben sich nicht an der Fragebogenaktion beteiligt, um dem Prozess keine zusätzliche Legitimation zu verleihen.
Zwar heißt es im Hintergrundpapier der GIZ: „Die Steuerungsstruktur des Global Health Hubs z.B. in Bezug auf Entscheidungsgremien, Mitglieder und Rollenverteilung soll in der Konzeptionsphase vorgeschlagen werden.“ Aber eine echte Debatte über Sinn und Zweck des Ganzen scheint nicht mehr möglich. Wie stark gesteuert der Prozess ist, zeigt auch ein Treffen am 16. November, zu dem sehr selektiv eingeladen wurde. TeilnehmerInnen des Treffens berichten von einer eher konfusen Veranstaltung. Die GIZ hatte aus den Fragebögen, getrennten Gesprächen mit den Interessengruppen und „besonderen Wissensträgern“ Ziele kondensiert. Die passten auf zwei Folien und sagen weniger aus als die im Oktober zirkulierte Beschreibung (siehe Kasten). Viel Raum nahm dagegen die geplante Struktur des GHHG ein, die im Fragebogen gar nicht abgefragt worden war. Vielleicht haben dazu die „besonderen Wissensträger“ beigetragen. Deren Auswahl erscheint ziemlich selektiv: Ein Vertreter des World Health Summit, Christoph Benn vom Globalen Fonds, Herr Schmitz Guinote von der WHO, ein Vertreter des von der Bundesregierung 2017 gegründeten AMR-Hub, der Forschung gegen Antibiotika-Resistenzen forcieren soll, Ilona Kickbusch, Vorsitzende des Beratungsgremiums des Gesundheitsministeriums zu globaler Gesundheit und schließlich ein Vertreter des von Kickbusch geleiteten Global Health Centre in Genf. Die Zivilgesellschaft scheint offensichtlich nicht über „besonderes Wissen“ zu verfügen.
Das Treffen am 16. November verstärkte den Eindruck, dass die Struktur des Health Hub längst beschlossene Sache ist und es bestenfalls noch darum geht, wer mitmachen darf. Kritische Fragen wie denn der Anspruch des „Arbeitens auf Augenhöhe“ (GIZ) eingelöst werden solle angesichts der teils widersprüchlichen Ziele und Ressourcen der Akteure, fanden ebenso wenig eine befriedigende Antwort, wie Nachfragen nach der Notwendigkeit und Sinnhaftigkeit des Hub.
Gesteuert werden soll der GHHG von „10 bis 15 hochrangigen Vertretern verschiedener Akteursgruppen“, einschließlich nachgeordneter Behörden und der Bundesregierung. Geplante „Veranstaltungsformate“ wie „Kamingespräche“ , die in der Regel vertraulich ablaufen, lassen ein intransparentes Vorgehen erwarten.
Angesichts der massiven Repräsentanz von IndustrievertreterInnen in den Diskussionen könnte man zu dem Schluss kommen, dass das nach außen postulierte Ziel der Umsetzung des nachhaltigen Entwicklungsziels (SDG 3) „Gesundes Leben für alle“ gar nicht im Vordergrund steht, sondern eine Schadensbegrenzung im Sinne einer Sicherung wirtschaftlicher Interessen. Bei der Beschreibung der Aktivitäten des GHHG ist viel von Information, Austausch, Kooperation und „innovativen Ansätzen“ die Rede, nicht aber von der Einforderung konkreter politischer Veränderungen, um dem Ziel „Gesundheit für Alle“ näher zu kommen.
Vielleicht ist es kein Zufall, dass die GIZ das SDG 17 „Partnerschaften“ zuerst nennt. Wenn es die Industrie schafft, kritische Gruppen in einen Diskurs zu verstricken, der auf langwierige Kompromissfindungen hinausläuft oder die Akteure einfach nur mit Nebensächlichkeiten beschäftigt hält, hat sie ein wichtiges Ziel erreicht. Nach außen hin halten sich die Industrieakteure vornehm im Hintergrund, denn das Wichtigste ist, erst einmal mit im Boot zu sitzen. Da sie schon bei der Kiellegung dabei war, nicht weiter schwierig.
Das alles lässt Erinnerungen an die Entstehung des „Deutschen Netzwerk gegen vernachlässigte Tropenkrankheiten“ wachwerden. Das wurde von Verband forschender Arzneimittelunternehmen (Vfa) gegründet, schaffte es aber geschickt, wichtige Akteure mit an Bord zu holen.[4] Koordiniert wird das Netzwerk von g+h communication, einer PR-Agentur die auch für den Vfa tätig ist.
Finanzierung
Zweifelhaft ist auch die Finanzierung des GHHG. Die Bundesregierung übernimmt für drei Jahre eine Anschubfinanzierung von einer Mio. € pro Jahr. Anschließend sollen die Stakeholder die Finanzierung sichern. Wie heißt es so treffend: „Wer bezahlt, bestimmt die Musik“. Pikantes Detail am Rande: Im Ablaufplan steht die Entwicklung eines Logos für den GHHG zeitlich vor der Ausarbeitung eines inhaltlichen Konzeptpapiers.
Die entscheidende Frage aber bleibt: Wer braucht den GHHG überhaupt? NGOs, Gewerkschaften und Wissenschaft sind durch die Deutsche Plattform Globale Gesundheit bereits vernetzt, im Entwicklungshilfebereich gibt es VENRO, das Aktionsbündnis gegen Aids und den Arbeitskreis medizinische Entwicklungshilfe (AKME). Profitieren würde dagegen die Industrie, die den Diskurs über Globale Gesundheit damit ebenso elegant beeinflussen könnte wie auch die Gates Stiftung und der World Health Summit, die ihre jeweils eigene Agenda haben.
Während für den GHHG eingefordert wird, dass die TeilnehmerInnen „auf Augenhöhe“ und „partnerschaftlich“ zusammenarbeiten und der intersektorale Dialog und die Kooperation gefördert wird, scheint die Botschaft bei der Bundesregierung noch nicht so recht angekommen zu sein. Der GHHG ist ein Kind des Gesundheitsministeriums. Das Entwicklungshilfeministerium sitzt bestenfalls am Katzentisch dabei. Und das Forschungsministerium, auch nicht unwichtig für die Neuausrichtung, hat eine eigene „Plattform globale Gesundheit“ angekündigt.
Irritierend ist der GHHG auch aus einem anderen Grund: Die Bundesregierung diskutiert ihre globale Gesundheitsstrategie derzeit noch, sie soll erst im Laufe des kommenden Jahres verabschiedet werden. Dazu gibt es einen begleitenden Prozess, bei dem verschiedene Interessenträger mitdiskutieren können.
Der GHHG soll schon im Januar 2019 seine Arbeit aufnehmen. Welchen Sinn hat diese Parallelstruktur, wenn nicht privilegierten Gruppen die exklusive Möglichkeit zu bieten, die Debatte in eine ihr genehme Richtung zu lenken und politische Schritte, die ihren Interessen schaden, zu verhindern oder mindestens zu verwässern? (JS)
Artikel aus dem Pharma-Brief 8-9/2018, S.1
[1] Pharma-Brief (2018) Bundesregierung hört zu. Nr. 7, S. 1
[2] GIZ (2018) Global Health Hub. Konzeptpapier Stand 1. Okt.
[3] Formell ist der Vfa kein eigens geladener Akteur, aber bei Treffen trotzdem stets gut vertreten. Im vom BDI gegründeten „German Health Care Partnership“ sind unter anderen einige Pharmafirmen Mitglied, die auch dem Vfa angehören.
[4] Pharma-Brief (2013) Pharmaindustrie erfindet die Zivilgesellschaft neu. Nr. 10, S. 6