
2021-artikel
Ziemlich illegal
Strafen für Big Pharma in den USA
Illegale Aktivitäten gehören offensichtlich zum Geschäftsmodell großer Pharmafirmen. 85% der 26 größten Firmen mussten über einen Zeitraum von 14 Jahren Strafzahlungen leisten, die meisten gleich mehrfach.[1]
Die Zahlen für den Zeitraum 2003-2016, die drei WissenschaftlerInnen aus den USA zusammentrugen, sind beeindruckend.[2] Doppelter Spitzenreiter war die Firma GlaxoSmithKline mit 27 Verstößen und knapp 10 Milliarden US$ an Strafen. Dennoch machten die Bußen nur 1,55% des Firmenumsatzes im gleichen Zeitraum aus. Unter den 22 Firmen, die es mit dem Gesetz nicht so genau nahmen, finden sich auch zwei Firmen mit Sitz in Deutschland: Bayer mit 13 Verstößen und 603 Mio. US$ Bußgeldern sowie Boehringer Ingelheim mit 7 Verstößen und 416 Mio. US$ Bußen.
 Häufigster Grund für Gerichtsverfahren waren überhöhte Preise für Medikamente, die durch staatliche Programme finanziert wurden (78 Fälle). 50 Verfahren richteten sich gegen Vermarktung für nicht zugelassene Indikationen. Schmiergelder (Kickbacks) zur Umsatzsteigerung (33) und irreführende Vermarktungspraktiken (32) kamen etwa gleich häufig vor. Je 21-mal führten das Verschweigen von negativen Informationen und Umweltvergehen zu Verfahren, die in Bußgelder mündeten. Weitere Gründe waren Bestechung (9), Finanzvergehen (Steuerbetrug oder Insiderhandel, 7-mal) sowie fünf Fälle von Qualitätsmängeln oder der Verkauf von nicht zugelassenen Produkten.
Häufigster Grund für Gerichtsverfahren waren überhöhte Preise für Medikamente, die durch staatliche Programme finanziert wurden (78 Fälle). 50 Verfahren richteten sich gegen Vermarktung für nicht zugelassene Indikationen. Schmiergelder (Kickbacks) zur Umsatzsteigerung (33) und irreführende Vermarktungspraktiken (32) kamen etwa gleich häufig vor. Je 21-mal führten das Verschweigen von negativen Informationen und Umweltvergehen zu Verfahren, die in Bußgelder mündeten. Weitere Gründe waren Bestechung (9), Finanzvergehen (Steuerbetrug oder Insiderhandel, 7-mal) sowie fünf Fälle von Qualitätsmängeln oder der Verkauf von nicht zugelassenen Produkten.
Die Verstöße waren meist kein kurzer Fehltritt, im Mittel dauerten sie je nach Firma zwischen 4 und 11 Jahre an. Einzige Ausnahme war Perrigo, die nur einen Verstoß zu verzeichnen hatte, der sich über ein Jahr erstreckte.
Nur bei vier Firmen machten die Bußen mehr als 1% des Umsatzes aus, Spitzenreiter mit 2,05% war Schering-Plough.[3]Angesichts von wiederholten Verstößen gegen gesetzliche Regeln drängt sich der Verdacht auf, dass Strafen einfach eingepreist werden. Schon seit einiger Zeit gibt es in den USA deshalb die Forderung, die Bußen zu erhöhen und schwere Verstöße mit Haftstrafen für die verantwortlichen ManagerInnen zu belegen. (JS)
Artikel aus Pharma-Brief 1/2021, S. 5
Bild Dollarscheine © vitpho/iStock
[1] Bei vier Firmen wurden im untersuchten Zeitraum keine Verstöße gefunden: Biogen Idec, Celgene, Gilead Sciences und Hospira.
[2] Arnold DG et al. (2020) Financial Penalties Imposed on Large Pharmaceutical Firms for Illegal Activities JAMA; 324, p 1995
[3] Inzwischen von Merck &Co aufgekauft.
WTO-„Waiver“: Genfer Grabenkämpfe
Alle zwei Jahre tagt üblicherweise das höchste Entscheidungsgremium der Welthandelsorganisation (WTO). Das jüngste Treffen der MinisterInnen-Konferenz war ursprünglich für Juni 2020 in Kasachstan geplant, findet aber stattdessen mit über einem Jahr Verspätung von Ende November bis Anfang Dezember in der Schweiz statt. Es böte die Gelegenheit, einen Befreiungsschlag beim Thema „Patent Waiver“ – also dem temporären Verzicht auf geistige Eigentumsrechte für Covid-19 Produkte – herbeizuführen. Nach mehr als dreizehn Monaten Debatte und rücksichtsloser Blockade einiger weniger Staaten, darunter auch Deutschland (wir berichteten[1]), ist jedoch unklar, wie noch zeitnah Bewegung in den festgefahrenen Prozess kommen könnte. Und das angesichts einer grotesken Ungleichheit in der globalen Verteilung von Covid-19-Impfstoffen.
Gegner des Waiver-Entwurfs setzen auf das Anziehen der globalen Impfstoff-Produktion in den kommenden Monaten und die Möglichkeit von Zwangslizenzen. Entsprechende EU-Vorschläge bleiben allerdings in den Augen kritischer Betrachterinnen weitgehend nutzlos.[2] Zuletzt zeigte sich die norwegische Verhandlungsführung im TRIPS Council offenbar frustriert vom lähmenden Status-quo.[3] Dennoch finden sich auch Meldungen mit etwas optimistischeren Einschätzungen, etwa einer angeblichen Annäherung zwischen Südafrika und der EU.[4] Offenbar versucht die WTO-Führung zudem, durch die Verabschiedung eines „Gesundheitspakets“ bei der MinisterInnen-Konferenz, wichtige Staaten wie Indien zur Zustimmung bei anderen umkämpften Themen zu gewinnen, etwa dem Ringen um ein Fischereiabkommen.[5] Das Treffen in Genf wird zeigen, ob sich die WTO überhaupt einen Rest von Handlungsfähigkeit bewahrt hat. (MK)
Artikel aus dem Pharma-Brief 8-9/2021, S. 5
[1] Pharma-Brief (2021) WTO: „Waiver“-Streit spitzt sich zu, Nr. 6, S. 8
[2] ‚t Hoen E & Boulet P (2021) The EU proposed Covid waivers of certain TRIPS rules are mostly meaningless. https://medicineslawandpolicy.org/2021/10/the-eu-proposed-covid-waivers-of-certain-trips-rules-are-mostly-meaningless/ [Zugriff 20.10.2021]
[3] Farge E (2021) A year after Covid vaccine waiver proposal, WTO talks are deadlocked. Reuters 4 Oct. www.reuters.com/article/us-health-coronavirus-wto-idAFKBN2GU1X7 [Zugriff 20.10.2021]
[4] Blenkinsop P et al. (2021) EU, South Africa hold „intense talks“ to break vaccine patent impasse. www.reuters.com/business/healthcare-pharmaceuticals/eu-south-africa-hold-intense-talks-break-vaccine-patent-impasse-2021-10-14/ [Zugriff 20.10.2021]
[5] The Times of India (2021) India, S Africa ask EU to break deadlock on Covid drugs, vax. 8 Nov. https://timesofindia.indiatimes.com/business/india-business/india-s-africa-ask-eu-to-break-deadlock-on-covid-drugs-vax/articleshow/87575225.cms [Zugriff 8.11.2021]
WHO Pandemie-Vertrag – Fortschritt oder Placebo?
Eine Sondersitzung der Weltgesundheitsversammlung hat den Auftrag zur Erstellung eines Pandemievertrags erteilt. Damit soll die Weltgesundheitsorganisation (WHO) bessere Instrumente an die Hand bekommen, um auf Pandemien reagieren zu können. Aber bis dahin ist der Weg noch weit und voller Klippen.
Die WHO kann auch bislang schon durch die Internationalen Gesundheitsvorschriften (IHR) bei Krisen weltweit intervenieren (siehe Kasten). Diese Vorschriften haben sich aber als nicht ausreichend erwiesen, wie die jüngste Covid-19-Pandemie zeigt. Denn die IHR enthalten keine Bestimmungen über den weltweiten Zugang zu Präventions- und Behandlungsmaßnahmen. Dem soll ein noch zu schreibender Pandemievertrag abhelfen. Das hat die Weltgesundheitsversammlung (WHA) am 1.12.2021 beschlossen. Ziel soll „ein umfassender und kohärenter Ansatz zur Stärkung der globalen Gesundheitsarchitektur“ sein, der neue Instrumente zur Pandemieprävention, der Vorbereitung für den Ernstfall und der angemessenen Reaktion entwickelt „und dabei die Priorität auf Gerechtigkeit setzt.“[1]
Langzeitprojekt
Bis März 2022 soll ein zwischenstaatliches Verhandlungsgremium (Intergovernmental Negotiating Body, INB) mit einer fairen Beteiligung von reichen und armen Ländern und allen Weltregionen gegründet werden. Zwischenergebnisse sollen bei der WHA im Mai 2023 vorgestellt werden, und bei der WHA 2024 soll der fertige Entwurf vorliegen. Welche Form das Vertragswerk annehmen soll, ist dabei noch offen. Es kann also eine WHO-Konvention werden (wie die zur Tabakkontrolle), eine Ergänzung der Internationalen Gesundheitsvorschriften oder ein Vertrag außerhalb der WHO.
Lob und Kritik
Dass die Gerechtigkeitsfrage gestellt wird, ist sicher als Fortschritt anzusehen. Und entgegen ursprünglichen Befürchtungen ist die Koordination bei der WHO angesiedelt und Hauptakteure sind die Mitgliedsstaaten.
Neben Lob für das Vorhaben gab es auch Kritik. Dass Industrieländer, die den Zugang zu Impfstoffen blockieren, sich für den Pandemievertrag stark gemacht haben, wirft Fragen nach der Ernsthaftigkeit auf.[2] Das People’s Health Movement sieht die Gefahr, dass es sich auch um eine Verzögerungstaktik handeln könnte.[3] Nach einigem Zögern haben allerdings viele Staaten die WHO-Resolution unterstützt.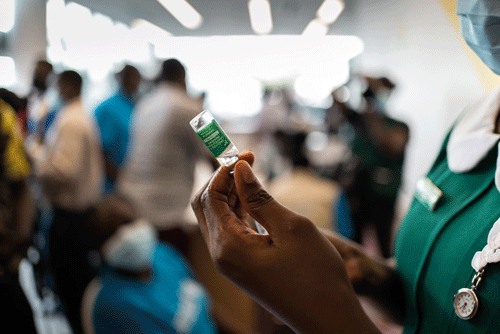
Auch die Gefahr eines Stakeholder-Ansatzes ist nicht zu unterschätzen: Die Resolution sieht vor, dass der INB weitere Akteure in die Verhandlungen mit einbeziehen kann. Also könnten auch kommerzielle Interessenträger und demokratisch nicht kontrollierte Stiftungen Einfluss auf die Debatte bekommen. Auch da gilt es aufmerksam zu bleiben – vor allem, weil außer der generellen Zielsetzung im Grunde noch alles offen ist.
Und was nicht in Vergessenheit geraten darf: Der geplante Vertrag ist kein Ersatz für akut notwendige Maßnahmen zur Bewältigung der Covid-19 Krise. Und wie sollen Staaten einer Pandemie überhaupt wirkungsvoll begegnen, wenn oft schon der Alltag in der regulären Versorgung eine permanente Katastrophe ist? Gerechtigkeit muss jetzt her. (JS)
Was geht jetzt schon?
Die WHO kann auch bislang schon durch die zuletzt 2005 aktualisierten Internationalen Gesundheitsvorschriften (International Health Regulations, IHR)[4] bei grenzüberschreitenden Gesundheitsbedrohungen aktiv werden. Die IHR sind ein völkerrechtlich bindendes Vertragswerk. Mitgliedsstaaten sind verpflichtet, geeignete Überwachungssysteme aufzubauen und potenzielle Bedrohungen zu melden. Sie müssen Strukturen etablieren, mit denen Gesundheitskrisen gemeistert werden können. Wenn die WHO einen Gesundheitsnotstand ausruft, kann sie Beschränkungen für den internationalen Verkehr von Menschen und Gütern erlassen sowie Eindämmungsmaßnahmen für alle Länder vorschlagen.
Artikel aus dem Pharma-Brief 10/2021, S.3
Bild © WHO/Blink Media - Nana Kofi Acquah
[1] WHA (2021) The World Together: Establishment of an intergovernmental negotiating body to strengthen pandemic prevention, preparedness and response. SSA2(5) 1 Dec https://apps.who.int/gb/ebwha/pdf_files/WHASSA2/SSA2(5)-en.pdf
[2] Karunakara U (2021) Europe Cannot ‘Treaty’ its Way Out of the Pandemic. Health Policy Watch. 30 Nov https://healthpolicy-watch.news/europe-treaty-pandemic/
[3] Rhodes N (2021) Do we need a pandemic treaty now? People’s Health Movement. https://phmovement.org/do-we-need-a-pandemic-treaty-now-policy-brief-by-peoples-health-movement/ [Zugriff 1.12.2021]
[4] WHO (2005) International Health Regulations. https://apps.who.int/iris/bitstream/handle/10665/246107/9789241580496-eng.pdf
USA: Zulassung ohne Evidenz
Alzheimer-Medikament trotz Kritik zugelassen
Das Alzheimer-Mittel Aducanumab wurde in den USA im Juni 2021 trotz scharfer Proteste von WissenschaftlerInnen[1] zugelassen. Eine fragwürdige Entscheidung mit gleich mehreren negativen Folgen.
Die Firma Biogen hatte zwei Studien zu dem Wirkstoff durchgeführt, die beide im März 2019 wegen Unwirksamkeit abgebrochen worden waren.[2] Erst durch eine spätere Auswertung einer Teilgruppe in einer Studie fand sich ein schwacher Effekt.[1] Solche nachträglichen Analysen können eigentlich niemals als Beweis einer Wirksamkeit gelten, sondern bestenfalls Hypothesen für weitere Untersuchungen generieren.
Deshalb hatte in der Anhörung bei der US-Zulassungsbehörde FDA auch keiner der ExpertInnen für eine Zulassung gestimmt, die allermeisten sogar explizit dagegen. Drei davon äußerten ihr Unverständnis sogar öffentlich in der Fachzeitschrift JAMA.[3] Einer der Berater, Caleb Alexander Johns von der Hopkins Bloomberg School of Public Health, bezeichnete die nachträgliche Analyse der Daten als „Texas sharpshooter fallacy“: Ein Scharfschütze zielt auf eine Scheunenwand und malt anschließend das Zielkreuz um die Stelle mit den meisten Treffern.[2]
Aller Protest half nichts. Die FDA ließ Aducanumab am 7. Juni 2021 zu und das sogar in einem beschleunigten Verfahren. Letztlich wurde ein Unterschied bei den beta-Amyloid-Ablagerungen als entscheidend angesehen. Das ist ein Surrogat-Marker, der umstritten ist. Zwar wurden in einer Phase 1 Studie Unterschiede zugunsten von Aducanumab gefunden, aber in den Phase 3 Studien gab es keinen klaren Zusammenhang zwischen den Ablagerungen und der klinischen Entwicklungen bei den PatientInnen.[4]
Über zwei Dutzend Studien mit anderen Wirkstoffen konnten keinen eindeutigen Zusammenhang zwischen Amyloid und dem Fortschreiten der Demenz ermitteln. Manche Wirkstoffe senkten den Wert und der Zustand der PatientInnen verschlechterte sich, bei anderen passierte gar nichts. Zwei Experten kommentierten fassungslos: „Ob beta-Amyloid allein ein valides Surrogat für die Behandlung von Alzheimer ist, ist unklar, und war bis zum Morgen des 7. Juni 2021 ein Gegenstand von andauernden und wichtigen Untersuchungen. Jetzt ist die Behandlung des Amyloids plötzlich klinische Praxis.“[4]
Risiken
So wenig der Nutzen des Medikaments belegt ist, so sicher ist, dass es nicht unwesentliche Nebenwirkungen verursacht. Bei 35 von 100 PatientInnen kommt es zu Gehirnschwellungen, die bei einem kleineren Teil der Betroffenen zu Kopfschmerzen, Verwirrtheit oder Schwindel führen. Mikroblutungen im Gehirn treten bei 19 von 100 auf. Beide Nebenwirkungen treten deutlich häufiger auf als unter Placebo. PatientInnen müssen sich während der Behandlung unangenehmen und teuren MRT-Untersuchungen unterziehen. Alle vier Wochen ist eine einstündige Infusion von Aducanumab nötig[5] – eine nicht unbedeutende Einschränkung der Lebensqualität.
Projekt Onyx
BeobachterInnen haben gerätselt, wie eine solche – wissenschaftlich nur schwer erklärbare – Entscheidung der FDA zustande kommen konnte. Das bekannte Pharma-Informationsportal STAT hat nach umfangreichen Recherchen eine plausible Erklärung gefunden.[6] Biogen hatte einen direkten Zugang zu Billy Dunn, dem für Alzheimer zuständigen Wissenschaftler in der Behörde. Die Firma startete vor zwei Jahren das geheime „Projekt Onyx“, um dem bereits gescheiterten Produkt neues Leben einzuhauchen. Zwar war bekannt, dass es eine Art zweifelhafter Kooperation zwischen der FDA und Biogen gab, aber Treffen fanden bereits viel früher statt als die FDA bisher einräumte. Bei einer Neurologen-Konferenz traf sich Biogen-Chefwissenschaftler Sandrock mit Dunn und erzählte ihm von doch noch entdeckten vorteilhaften Ergebnissen. Das war ein klarer Verstoß gegen die Regeln der Behörde. Denn Firmen dürfen ihre Studiendaten der FDA nur ein einem klar definierten Setting mitteilen, um Mauscheleien und Beeinflussungsversuche zu unterbinden.
Eichlers Traum
Es besteht die akute Gefahr, dass Aducanumab zum Präzedenzfall für weitere zweifelhafte Alzheimer-Produkte wird. Es sieht so aus, dass der Traum von Hans-Georg Eichler, dem ehemaligen Chef für Humanarzneimittel bei der europäischen Arzneimittelbehörde EMA, zur Realität wird. Eichler hatte sich über viele Jahre für schnellere Zulassungen auf Basis von Surrogatendpunkten eingesetzt, um die Industrie von Kosten zu entlasten.[7] In einem Papier von 2015 hatte er genau Alzheimer-Medikamente als Begründung genannt. Es sei den Herstellern nicht zuzumuten, 15-20 Jahre lang Studien durchzuführen, bis man sehen könne, ob die Medikamente tatsächlich nützten. Und das, obwohl er selbst einräumt, dass die Amyloid-Hypothese nicht gesichert ist.[8]
Genau das trifft auf Aducanumab zu: Ergebnisse der Phase 4-Studie werden nicht vor 2030 erwartet. Bei Jahreskosten von 56.000 US$ pro Jahr plus teuren notwendigen MRTs ein schlechtes Geschäft. Selbst unter der Annahme, dass die behaupten geringen Effekte real wären, kommt das pharmaökonomische Institut ICER zu dem Ergebnis, dass nur ein Preis von 3.000 bis 8.400 US$ gerechtfertigt wäre.[9] Das alles bedeutet aber, dass die Studien nach der Zulassung auf Kosten und Risiko der Allgemeinbevölkerung durchgeführt werden. (JS)
Artikel aus dem Pharma-Brief 6/2021, S.3
[1] Pharma-Brief (2021) USA: Wie wenig Evidenz darf’s sein? Nr. 3-4, S. 8
[2] Boseley S (2021) FDA approves first new Alzheimer’s drug in almost 20 years. Guardian 7 June www.theguardian.com/society/2021/jun/07/fda-announce-decision-new-alzheimers-drug-aducanumab
[3] Alexander C et al. (2021) Evaluation of Aducanumab for Alzheimer Disease. Scientific Evidence and Regulatory Review Involving Efficacy, Safety, and Futility. JAMA; 325, p 1717
[4] Alexander CG and Karlawish J (2021) The Problem of Aducanumab for the Treatment of Alzheimer Disease. Ann Int Med https://doi.org/10.7326/M21-2603
[5] FDA (2021) Label Aducanumab www.accessdata.fda.gov/drugsatfda_docs/label/2021/761178s000lbl.pdf (Abruf 19.6.2021)
[6] Feuerstein et al. (2021) Inside ‘Project Onyx’: How Biogen used an FDA back channel to win approval of its polarizing Alzheimer’s drug. Boston Globe. 29 June
[7] Pharma-Brief (2016) Pilotprojekt gescheitert –weiter so? Nr. 7, S: 1
[8] Eichler HG et al. (2015) From Adaptive Licensing to Adaptive Pathways: Delivering a Flexible Life-Span Approach to Bring New Drugs to Patients. Clinical Pharmacology & Therapeutics; 97 p 234
[9] ICER (2021) In Revised Evidence Report, ICER Confirms Judgment That Evidence is Insufficient to Demonstrate Net Health Benefit of Aducanumab for Patients with Alzheimer’s Disease. Press release 30 June https://icer.org/news-insights/press-releases/in-revised-evidence-report-icer-confirms-judgment-that-evidence-is-insufficient-to-demonstrate-net-health-benefit-of-aducanumab-for-patients-with-alzheimers-disease [Zugriff 21.7.2021]
Südafrika: Bei HIV, TB und Krebs schlechter versorgt
Pandemie und Lockdown brachten massive Einschränkungen
Südafrika hatte von allen afrikanischen Ländern mit Abstand die höchsten Infektionsraten und Todesfälle durch COVID-19. Fast 90.000 Menschen starben, trotz rigider Lockdown-Maßnahmen.[1] Viele Bereiche der Gesundheitsversorgung wurden durch die Pandemie eingeschränkt, etwa die Behandlung und Früherkennung von TB, HIV oder Krebs. Stark beeinträchtigt waren auch Dienste für sexuelle und reproduktive Gesundheit.
Südafrika war und ist von der Pandemie schwer betroffen. Nachdem am 5. März 2020 der erste Krankheitsfall durch SARS-CoV-2 bekannt geworden war, stieg die Infektionskurve rasant. Bereits am 27. März verhängte die Regierung „einen der striktesten Lockdowns außerhalb Chinas“.[2] Neben der Schließung von Schulen, Universitäten und Geschäften gab es strenge Ausgangs- und Reisebeschränkungen. SüdafrikanerInnen konnten nicht einmal mehr ihren Hund spazieren führen, öffentliche Transportmittel benutzen oder im Freien Sport treiben. Das Haus verlassen durfte nur, wer Lebensmittel, Medikamente oder Kraftstoffe zum Heizen besorgen wollte. Ohne öffentliche Verkehrsmittel hatten arme Menschen jedoch kaum eine Chance, Gesundheitsdienste zu erreichen. Erst nach gut einem Monat gab es nach und nach Lockerungen.
Lockdown trifft besonders die Armen
Chitsamatanga und Malinga, die die Situation analysierten, kritisieren vor allem, dass die Regierung einen „One size fits all“ Ansatz verfolgte, wodurch marginalisierte Bevölkerungsgruppen besonders stark unter den Auswirkungen der Pandemie litten. Maßnahmen wie Ausgangsbeschränkungen, Schul-, und Geschäftsschließungen, die in westlichen Ländern gut funktionieren, können in einem Land in dem rund 3 Millionen Menschen im informellen Sektor arbeiten und von „der Hand in den Mund“ leben, nicht eins zu eins übernommen werden.[2]
Schon vor der Pandemie gab es große Unterschiede bei der Qualität der Versorgung: So kommt im privaten Sektor ein Arzt auf etwa 500 Versicherte, im öffentlichen Sektor muss eine Ärztin dagegen 2.500 Menschen versorgen.[3] Weniger als 20% der Bevölkerung sind jedoch privat krankenversichert und können sich eine solche Vorzugsbehandlung leisten. Der strikte Lockdown verschärfte die Ungleichheiten und blockierte den Zugang zu vielen Bereichen der Gesundheitsversorgung.
HIV und TB: Versorgung blieb auf der Strecke
Zum Beispiel HIV und Tuberkulose: Südafrika zählt zu den „Three High Burden“ Ländern, also zu den 20 Ländern mit der höchsten Krankheitslast an HIV/AIDS, Tuberkulose sowie multiresistenter TB.[4] Wegen der Covid-19 Pandemie wurden etliche Präventionsangebote und Versorgungsleistungen für die Betroffenen eingeschränkt oder ganz ausgesetzt. Zwar ergab eine Untersuchung unter 65 Primärkliniken, dass die Behandlungsprogramme für Personen, die bereits eine antiretrovirale Therapie (ART) erhielten, größtenteils aufrecht erhalten wurden. Es gab aber mit Beginn des Lockdowns einen deutlichen Rückgang bei den HIV-Testungen (um 48%). Die Zahl der HIV-Infizierten, die eine Behandlung starteten, reduzierte sich im April 2020 ebenfalls um fast die Hälfte (46%). Zwar hat sich die Lage später wieder verbessert, doch im Juli 2020 waren noch immer nicht die Test- und Behandlungszahlen aus dem Zeitraum vor der Pandemie erreicht. Die Unterbrechung beim Testen und der vielfach verzögerte Behandlungsbeginn werde die HIV-Infektionen in die Höhe treiben, schätzen ExpertInnen. Allein zwischen Juli und September 2020 sind geschätzt 11.000 HIV-Infektionen unentdeckt geblieben. Dadurch sei auch ein Anstieg von HIV-Infektionen bei Neugeborenen zu erwarten.[5]
Bei Tuberkulose kam es aufgrund der Pandemie ebenfalls zu starken Einschränkungen. TB-Stationen wurden zum Teil in Covid-19 Stationen umgewandelt und Personal wurde zur Testung und Behandlung des SARS-CoV-2 Virus abgezogen. Laut den Laboren des staatlichen Gesundheitssystems haben die Einschränkungen der höchsten Lockdown-Stufe 5 im ersten Pandemiemonat zu einem Rückgang der Anzahl von TB-Tests um 48% geführt. 33% weniger Tuberkulose-Fälle als zuvor wurden diagnostiziert.[6]
Verhütungsmittel wurden knapp
Mehr als zwei Drittel aller Südafrikaner beziehen Kondome aus öffentlichen Gesundheitseinrichtungen, wo sie kostenlos verteilt werden. Die Schließung vieler Stationen und der landesweite Lockdown machten das unmöglich. In einer Studie gaben knapp 23% der Befragten an, in dieser Zeit keinen Zugang zu Kondomen gehabt zu haben.6 Das dürfte nicht nur die Anzahl ungewollter Schwangerschaften sondern auch von sexuell-übertragbaren Erkrankungen in die Höhe getrieben haben.
Nicht einmal 10% der SüdafrikanerInnen, die verhüten, nutzen die Pille. Langzeitkontrazeptiva werden häufig genutzt, doch auch sie waren schwerer zugänglich. In der Provinz Gauteng wurden während des Lockdowns 48% weniger Hormonimplantate eingesetzt und 10% weniger Intrauterinpessare (IUCD).[7] Das Gesundheits-JournalistInnen Netzwerk Health-e News berichtet zudem über gravierende Versorgungsengpässe bei Verhütungsspritzen im Juli 2020.[8] Zudem wurden während der Pandemie wesentlich seltener Abtreibungen durchgeführt, Frauen wurden von den Kliniken abgewiesen.[9] Das wird höchst wahrscheinlich zur vermehrten Nutzung riskanter Abtreibungspraktiken geführt haben.[10]
In der bevölkerungsreichsten Provinz Guateng ist die Anzahl der Geburten durch minderjährige Mütter um 60% gestiegen. 2019 brachten 14.577 unter 18jährige Frauen ein Kind zur Welt – zwischen April 2020 und März 2021 waren es 23.000.[11] In einer Befragung von 15 bis 25-jährigen Frauen und Mädchen gaben etwa 14% an, dass seit der Pandemie mehr Gewalt in ihrem Zuhause herrscht. Das Risiko und die Angst vor körperlicher und seelischer Gewalt sowie sexuellen Missbrauchs sei durch die Pandemie deutlich gewachsen.[12]
KrebspatientInnen: schlechter versorgt als zuvor
Bei der Versorgung von PatientInnen mit nicht-übertragbaren Krankheiten (NCDs) gab es bedingt durch die Pandemie ebenfalls gravierende Einschnitte. Zum Beispiel bei Krebs: In 28 % der untersuchten Krankenhäuser in Südafrika wurden Tumor-Operationen ganz gestrichen oder reduziert. Auch Vorsorgemaßnahmen wurden eingeschränkt und es gab u.a. deutlich weniger Screenings oder HPV-Impfungen zum Schutz vor Gebärmutterhalskrebs[13]. Weil MitarbeiterInnen von onkologischen Stationen abgezogen bzw. bei der Versorgung von COVID-PatientInnen eingesetzt wurden, konnten Versorgungsleistungen für Krebs-PatientInnen zum Teil nicht mehr angeboten werden. Hinzu kam ein Mangel an Krebsmedikamenten und anderen wichtigen Medizinprodukten aufgrund unterbrochener Lieferketten, der Schließung der Landesgrenzen und einer verringerten Produktion. Nicht zuletzt die Krebsforschung wurde in Südafrika erheblich zurückgefahren.[14]
Mit unserem Projekt „Globale Folgen der Pandemie“[15] werden wir weiterhin im Auge behalten wie sich die Lage der Gesundheitsversorgung in Südafrika und anderen Ländern entwickelt. Mehr dazu können Sie im kommenden Frühjahr auch in einem Pharma-Brief Spezial lesen. (SB/CJ)
Artikel aus dem Pharma-Brief 10/2021, S.4
Straßenkreuzung STAY HOME © Discott K
[1] John Hopkins University of Medicine (2021) Global Map. Coronavirus resource center https://coronavirus.jhu.edu/map.html [Zugriff 23.11.21]
[2] Chitsamatanga, B and Malinga, W (2020) ‘A tale of two paradoxes in response to COVID-19’: Public health system and socio-economic implications of the pandemic in South Africa and Zimbabwe. Cogent Social Sciences 7 (1869368), p 1-19
[3 ]Medical Brief (2018) Africa Check puts together the numbers on doctor-patient ratios. www.medicalbrief.co.za/africa-check-puts-together-numbers-doctor-patient-ratios/ [Zugriff 23.11.21]
[4] WHO (2020) Global Tuberculosis Report 2020. Geneva
[5] Dorward J et al. (2021) The impact of the COVID-19 lockdown on HIV care in 65 South African primary care clinics: an interrupted time series analysis. Lancet HIV 8, p e158
[6] USAID (2020) USAID/South Africa Tuberculosis South Africa Project (TBSAP) Midterm Evaluation Report. Silver Spring
[7] Bolarinwa, O (2021) Factors associated with limited access to condoms and sources of condoms during the COVID-19 pandemic in South Africa. medRxiv, p. 1-19. doi: https://doi.org/10.1101/2020.09.11.20192849
[8] Health-E News (2021) Contraception in SA: What you need to know https://health-e.org.za/2021/09/27/contraception-in-sa-what-you-need-to-know/ [Zugriff 2.12.21]
[9] Aussage von Bibi-Aisha Wadvalla / Managing Director von Health E-News im Gespräch mit der BUKO Pharma-Kampagne im August 2021
[10] Adelekan T et al. (2020) Early Effects of the COVID-19 Pandemic on Family Planning Utilisation and Termination of Pregnancy Services in Gauteng, South Africa: March–April 2020. Wits Journal of Clinical Medicine; 2, p 145
[11] 2019: 14.577; April 2020 - März 2021: 23.000Save the children (2021) Teen pregnancies in South Africa jump 60% during COVID-19 pandemic. https://reliefweb.int/report/south-africa/teen-pregnancies-south-africa-jump-60-during-covid-19-pandemic [Zugriff 23.11.21]
[12] Mathews C. et al. (2021): HerStory 2: Process evaluation of the combination HIV prevention intervention for adolescent girls and young women (AGYW), Global Fund grant period 2019 to 2022. Report 1/5: Overview of findings and combined recommendations, 1-92.
[13] Nnaji, C.; Moodley, J. (2021): Impact of the COVID-19 pandemic on cancer diagnosis, treatment and research in African health systems: a review of current evidence and contextual perspectives. Ecancer Medical Sciences 15 (1170), p. 1-10
[14 ]Addai, B.; Ngwa, W. (2021): COVID-19 and cancer in Africa. Sciences 371 (6524), p. 25-27. DOI: 10.1126/science.abd1016
[15] https://bukopharma.de/de/globale-folgen-der-pandemie
Pharma-Kampagne – One Health im Fokus
Arbeiten unter erschwerten Bedingungen: Jahresbericht 2020
Gesundheitskrisen erzeugen Handlungsdruck. Sie beschleunigen Innovationen und eröffnen Chancen für strukturellen Wandel. Das hat die Covid-19-Pandemie schmerzhaft bewiesen. Neben den vielen Schwierigkeiten, die uns das Corona-Jahr auferlegt hat, hat es zugleich auch Visionen und Ziele beflügelt: Die Rufe nach globalen Gesundheitsstrategien, einem gerechten Zugang zu innovativen Forschungsprodukten, nach internationaler und interdisziplinärer Zusammenarbeit wurden 2020 deutlich lauter.
Nicht zuletzt das One-Health-Konzept, das wir mit unserem Projekt zu Antibiotika-Resistenzen stark in den Fokus genommen hatten, gewann plötzlich eine neue Brisanz. Denn der größte Teil neuartiger Infektionskrankheiten stammt – wie auch Covid-19 – vom Tier. Die komplexen Zusammenhänge der Gesundheit von Tier, Mensch und Umwelt in den Blick zu nehmen, ist das Gebot der Stunde. Nur der Schulterschluss verschiedener Bereiche macht es möglich, neuartige oder auch resistente Krankheitserreger künftig besser eindämmen zu können.
Beim Thema Covid-19 hat sich die BUKO Pharma-Kampagne energisch für eine globale Sichtweise engagiert. Schon früh thematisierten wir wunde Punkte der Pandemiekontrolle und haben mit dafür gesorgt, dass der weltweite Zugang zu Impfstoffen ein wichtiges Thema in der öffentlichen Debatte wurde. Unsere neue Webseite zu Covid-19 präsentiert inzwischen die zahlreichen Artikel und Meldungen, die wir zum Thema veröffentlicht haben.
HIV: Kein Ende in Sicht!
Auch HIV/Aids stand weiterhin auf unserer Agenda. Denn Stigmatisierung, Kriminalisierung und hohe Preise schließen immer noch viele Menschen von Prävention und Behandlung aus. Unser neuer E-Learning-Kurs zeigt die vielfältigen Probleme mit gut verständlichen Texten und ansprechenden Fallbeispielen auf. Kurze Videos leiten jedes neue Kapitel ein und geben einen guten Überblick über die vielen Facetten des Themas. Der Kurs ging Mitte 2020 online und verzeichnete bis Jahresende erfreulicherweise bereits 732 Nutzungen. Waren Sie schon dort? Ein Besuch der informativen Webseiten lohnt sich und ist auch auf mobilen Endgeräten möglich! Nach Registrierung und Abschluss des kompletten Kurses können Sie ein Zertifikat erwerben.
Resistente Erreger:
Nicht zu stoppen?
Mit einem 64-seitigen Pharma-Brief Spezial zu Antibiotika-Resistenzen veröffentlichten wir im Frühjahr 2020 die Ergebnisse unserer Länderstudie zu Deutschland, Indien, Tansania und Südafrika. Die Broschüre steht in deutscher und englischer Sprache zur Verfügung. Sie liefert Daten zur Resistenzlage und beleuchtet die Ursachen und Folgen resistenter Erreger bei Menschen, Tieren und in der Umwelt. Die Länderberichte waren richtungsweisend für die Konzeption einer Präsentation: Unsere 16m2 große Wanderausstellung „Nicht zu stoppen“ bietet mit Stellwänden, Display-Säulen und Monitoren ungewohnte Perspektiven auf die Resistenz-Problematik in Süd und Nord. Im September wurde sie in Steinfurt erstmals öffentlich gezeigt, alle anderen Termine mussten jedoch wegen Covid-19 ausfallen. Sobald es die Infektionslage zulässt, wollen wir die Ausstellung 2021 erneut auf die Reise schicken.
Unsere gleichnamige Online-Ausstellung in völlig anderer Form ging im Oktober 2020 erfolgreich an den Start. Sie integriert zahlreiche Kurzfilme mit authentischen Berichten aus verschiedensten Teilen der Welt. Schauen Sie rein und geben Sie uns Ihr Feedback!
Viel Theater mit Corona
49 Auftritte, viel Sonnenschein und über 2.000 ZuschauerInnen machten unsere Theatertournee zu Antibiotika-Resistenzen trotz erschwerter Bedingungen zum vollen Erfolg. Die SchauspielerInnen überzeugten mit einem überwiegend pantomimischen Maskentheater. Doch nicht nur hier machte uns die Corona-Pandemie bzw. der erhebliche Mehraufwand zu schaffen. Mehr als ein Dutzend Veranstaltungen, die wir allein oder gemeinsam mit anderen Akteuren vorbereitet hatten, mussten entfallen oder verschoben werden. Andere Aktivitäten wurden komplett neu geplant oder abgewandelt, um sie Covid-19 kompatibel zu machen. All das kostete viel zusätzliche Mühe.
Viel Gehör gefunden
Trotzdem haben MitarbeiterInnen der Kampagne – digitaler Technik sei Dank – an über 50 Veranstaltungen teilgenommen. Die Pharma-Kampagne hat 13 Aufrufe und offene Briefe unterstützt und zudem eine äußerst erfolgreiche Pressearbeit geleistet: 51 JournalistInnen gaben wir Auskunft und insgesamt 65 Medienberichte thematisierten unsere Arbeit. Beeindruckend ist dabei die Bandbreite der Themen und Presseorgane: So führten wir Hintergrundgespräche mit Redakteuren des Spiegels, von ARD und ARTE zu diversen Themen mit Pharmabezug. Über die fehlende Transparenz bei klinischen Studien berichtete der Süddeutsche Rundfunk, das Deutsche Ärzteblatt, die Süddeutsche Zeitung und die Deutsche Apotheker Zeitung. Selbst die Apotheken-Umschau widmete der Thematik einen zweiseitigen Artikel.
Corona: Vernachlässigte Aspekte
Einen besonderen Schwerpunkt unserer Pressearbeit bildete gezwungenermaßen die Covid-19-Pandemie. Die Pharma-Kampagne veröffentlichte hierzu drei eigene Pressemitteilungen und über 100 Beiträge in sozialen Medien. Immer wieder war unsere kritische Einschätzung und Stellungnahme gefragt. Dem Evangelischen Pressedienst, Frontal 21, Report München oder auch dem NDR lieferten wir z.B. Hintergründe zur Covid-19-Forschung. Dem ARD Mittagsmagazin gaben wir Auskunft zu den Akteuren in der Pharmaforschung. Häufig kreisten Pressegespräche um Hürden des globalen Zugangs zu Impfstoffen, etwa bei WDR 5 oder der Deutschen Welle. Beiträge mit O-Tönen der Pharma-Kampagne brachten außerdem The Nation/USA, Times of Oman, Deutschlandfunk, Neues Deutschland oder die Zeitschrift Weltsichten.
Berichterstattung rund um unser Projekt zu Antibiotika-Resistenzen gab es besonders im Zeitraum unserer Theatertournee, die der Problematik größere Aufmerksamkeit verschaffte. Selbst in der Tagesschau kam die Pharma-Kampagne zu diesem Thema zu Wort.
Daneben waren auch hohe Preise für Krebsmedikamente ein Arbeitsbereich mit guter Presseresonanz: Das ARD Fernsehen plant dazu einen längeren Sendebeitrag und führte ein Interview mit Jörg Schaaber. Der FAZ lieferten wir Informationen und Zitate für den Artikel „Heilung um jeden Preis“.
Was gibt’s Neues?
Im vergangenen Jahr haben wir uns an die Suchmaschinen-Optimierung unserer Website herangetastet und werden hier auch 2021 am Ball bleiben. Unter anderem wird es demnächst größere Veränderungen auf unserer Startseite geben. Sie dürfen gespannt sein…
Und auch beim Pharma-Brief gibt es positive Neuerungen: Ende 2020 haben wir mit der Digitalisierung unserer Pharma-Brief-Artikel begonnen. Seit Ausgabe 7/2020 stehen nun alle Artikel auch einzeln online. Sie müssen nicht mehr umständlich eine PDF öffnen und bis zum gesuchten Beitrag blättern. Auch können die Beiträge nun leichter über Suchmaschinen gefunden oder von anderen Akteuren verlinkt werden. Für unsere Arbeit ist das ein großer Gewinn. Wir arbeiten daran, auch ältere Artikel nach und nach einfacher zugänglich zu machen.
Ohne die Unterstützung durch unsere Spenderinnen und Spender hätten wir all das nicht stemmen können! Auch die schnelle und unbürokratische Umwidmung von Geldern durch die Stiftung Umwelt und Entwicklung NRW hat sehr dazu beigetragen, dass wir unsere Projekte in diesem schwierigen Jahr erfolgreich durchführen konnten. Ein großes Dankeschön an alle, die unsere Arbeit kontinuierlich fördern und damit Veränderung möglich machen! (CJ)
Artikel aus Pharma-Brief 1/2021, S. 6
Bilder © Claudia Jenkes
Pfizers faule Covid-19 Impfstoff-Deals
Lateinamerikanische Staaten sollen für Fehler der Firma haften
Bei den rücksichtslosen Verhandlungen mit lateinamerikanischen Ländern pocht der Pharma-Gigant auf Schadensersatz auch bei selbstverschuldeten Problemen. Argentinien und Brasilien sollten sogar Staatsvermögen als Sicherheit einsetzen, um mögliche spätere Prozesskosten bezahlen zu können. In Peru dauerten die Verhandlungen fast sechs Monate. Im September 2020 wurde der Vertrag abgeschlossen. Er beinhaltet Klauseln, die die Firma bei Nebenwirkungen oder verspäteter Lieferung der Impfstoff-Chargen, teilweise von Schadensersatzforderungen freistellt.
Pfizer wird vorgeworfen, lateinamerikanische Länder bei den Covid-19 Impfstoff-Verhandlungen zu schikanieren. Mindestens in einem Fall haben die überzogenen Forderungen der Firma den Vertragsabschluss um drei Monate verzögert. Im Fall von Argentinien und Brasilien kam es am Ende zu keinem Vertragsabschluss.
Peru – Tödliche Folgen eines maroden Systems
Covid-19 offenbart Schwachpunkte der Versorgung
Covid-19 hat Peru schwer getroffen. Nirgendwo sonst ist die Sterberate so hoch:[1] Bis Mitte Juli wurden in dem südamerikanischen Land fast 200.000 Todesfälle verzeichnet – bei rund 33 Millionen EinwohnerInnen.[2] Es fehlt an ÄrztInnen, Intensivbetten und Sauerstoff. Andere Erkrankungen wie Malaria, die vor allem in der Amazonasregion endemisch ist, scheinen im Schatten der Pandemie zu verschwinden.
Patente sind auch keine Lösung
Warum wir andere Wege gehen müssen, um Forschung zu finanzieren
„Ohne Patente kein medizinischer Fortschritt“ – gerade in der Diskussion um den Patent-Waiver bei der Welthandelsorganisation wird Big Pharma nicht müde, dieses Argument zu wiederholen. Doch nicht nur hohe Arzneimittelpreise, die für große Teile der Weltbevölkerung unbezahlbar sind und fehlende Forschung für vernachlässigte Krankheiten strafen dieses Mantra Lügen. Auch die Tatsache, dass die entscheidende Grundlagenforschung vorwiegend öffentlich finanziert wird, macht ein Umdenken notwendig und wirtschaftlich attraktiv.
One World – One Health
Rückblick auf unsere ABR-Fachkonferenz
Der massive Verbrauch von Antibiotika fördert Resistenzen (ABR) und hat dramatische Auswirkungen auf die Gesundheit von Mensch, Tier und Umwelt. Alle drei Bereiche sind eng verknüpft. Daher ist entschlossenes Handeln gefragt, aber auch eine kohärente Politik. Eine zweitägige internationale Fachkonferenz der BUKO Pharma-Kampagne analysierte die gegenwärtigen Problemfelder und zeigte mögliche Handlungsstrategien auf.
Rund 160 Fachleute aus 10 Ländern diskutierten am 30.4. und 1.5.2021 auf Einladung der BUKO Pharma-Kampagne bei einer Online-Konferenz die globale Resistenzproblematik im Bereich Mensch, Tier und Umwelt. Ziel der Tagung war es, den internationalen und interdisziplinären Austausch zum Thema zu fördern, Lösungsansätze zu kommunizieren und damit auch Akteure aus der Politik zu erreichen.
„One World – One Health“ lautete der Konferenz-Titel und der Name war Programm: Gemäß dem One Health-Ansatz der WHO sollten Antibiotika-Resistenzen sektorübergreifend und global betrachtet werden. Neben ÄrztInnen, PharmazeutInnen und GesundheitswissenschaftlerInnen waren auch Fachleute aus der Veterinärmedizin, der Landwirtschaft und aus dem Umweltbereich zugegen.
Impulsvorträge aus vier Ländern
Fachleute aus verschiedenen Teilen der Welt waren am ersten Konferenztag zugeschaltet und präsentierten in ihren Keynotes kritische Analysen der lokalen Problemlage sowie Best Practice-Beispiele. Dieser internationale Konferenzteil fand in englischer Sprache statt.
Gopal Dabade, Arzt und Vorstand des All India Drug Action Network, richtete zunächst den Blick auf Indien. Er erläuterte die Zusammenhänge von Armut und Antibiotika-Resistenzen am Beispiel von Tuberkulose. Schlechte medizinische Versorgung, unzureichende Labortechnik und fehlende soziale Sicherungssysteme seien entscheidende Treiber der Resistenzentwicklung. Denn sie begünstigen Fehlbehandlungen durch informelle ÄrztInnen oder HeilerInnen sowie vorzeitige Therapieabbrüche wegen Geldmangels, so Dabade. Auch schlechte Wohnverhältnisse – insbesondere in städtischen Slums – forcieren die Ansteckung und die Ausbreitung von multiresistenten Erregern. Eine grundlegende Verbesserung der Lebensverhältnisse und der Zugang zu einer qualitativ hochwertigen Gesundheitsversorgung seien daher maßgeblich, um Resistenzen einzudämmen.
Andy Gray, Senior Lecturer für Pharmazie an der Universität KwaZulu-Natal in Durban, legte den Fokus auf den Antibiotika-Verbrauch in Südafrika. Hohe Verschreibungszahlen seien einerseits durch eine hohe HIV-Infektionsrate und damit einhergehenden bakteriellen Erkrankungen bedingt, andererseits Folge von Fehlverschreibung und -gebrauch. Als wirkungsvolle Maßnahme beschrieb er Südafrikas Antibiotic Stewardship Program, das in Krankenhäusern die Kommunikation zwischen MedizinerInnen, PharmazeutInnen und Pflegepersonal verbessert und damit rationale Therapieentscheidungen fördert.
Erick Venant, Pharmazeut und Gründer der Roll Back Antimicrobial Resistance Initiative in Tansania, berichtete ausführlich über Bildungsprojekte an Schulen und Social Media-Kampagnen als wirksames Instrument im Kampf gegen ABR. „Bildung ist der Schlüssel, um Antibiotika-Resistenzen effektiv zu bekämpfen“, so Venant.
Der Mikrobiologe und Infektions-Epidemiologe Dr. Gerhard Schwarzkopf-Steinhauser präsentierte Daten zur Resistenzlage in Deutschland. Er monierte u.a. ein lückenhaftes Resistenz-Monitoring. Es gebe zwar eine Verpflichtung zur Erfassung von Resistenzen, jedoch keine Meldepflicht der Labore. Nur rund ein Viertel der Krankenhäuser und ein Bruchteil der Arztpraxen seien an das Meldesystem des Robert Koch Instituts (RKI) angeschlossen.
Forderungen an die Politik
Im Anschluss an die Impulsvorträge wurden die Konferenzgäste durch eine Online-Ausstellung der BUKO Pharma-Kampagne zu Antibiotika-Resistenzen geführt. In drei Breakout-Räumen unternahmen sie eine virtuelle Reise durch die Themenbereiche Mensch, Tier und Umwelt. Kurzfilme und ReferentInnen aus verschiedenen Ländern lieferten jeweils Impulse für die Diskussion. Abschließend wurden in den Kleingruppen Ziele und Handlungsempfehlungen erarbeitet. Deutlich wurde dabei, dass beim Thema Antibiotika-Resistenzen bisher vor allem die Humanmedizin im Fokus steht, zunehmend auch die Tiermedizin. Das System Landwirtschaft mit seiner Massentierhaltung und erst recht die Umwelt werden jedoch weitgehend ignoriert und kommen in Aktionsplänen kaum vor. Das untergräbt nicht nur den One Health-Ansatz, es verzögert auch effektives und kohärentes Handeln.
Die in den Workshops aufgestellten politischen Forderungen flossen am nächsten Morgen in eine Podiumsdiskussion ein. Hier diskutierten drei Abgeordnete aus dem NRW Landtag – allesamt SprecherInnen ihrer Partei in Sachen Verbraucherschutz und Landwirtschaft – mit Bernhard Burdick von der Verbraucherzentrale NRW und dem Journalisten Christian Baars vom NDR, dessen Arbeitsschwerpunkt auf Gesundheitsthemen und Antibiotika-Resistenzen liegt. Die äußerst lebhafte Debatte thematisierte vor allem Landwirtschaft, Tierhaltung und Verbraucherschutz. Zur Sprache kamen etwa Dumpingpreise bei Fleischwaren, inakzeptable Haltungsbedingungen bei der Mast, fehlende Transparenz für VerbraucherInnen, Fragen des Gewässerschutzes sowie notwendige Systemveränderungen. Dass hier heiße Eisen angepackt werden müssen, zeigte sich auch im Chatverlauf sehr deutlich: Das Publikum lieferte zahlreiche Kommentare und Diskussionsbeiträge, die durch einen Anwalt des Publikums eingebracht wurden.
ABR – die globale Pandemie
Der zweite Konferenzteil lenkte den Blick stärker auf Deutschland. Er fand in deutscher Sprache statt und begann mit einem Impulsvortrag von Dr. Tim Eckmanns, Facharzt für Hygiene und Umweltmedizin und Leiter des Fachgebiets Nosokomiale Infektionen, Surveillance von Antibiotikaresistenz und -verbrauch am Robert Koch-Institut. Eckmanns erläuterte, welche Herausforderungen es in Deutschland und weltweit zu bewältigen gilt. Dabei verglich er die globale Ausbreitung von Antibiotika-Resistenzen mit der aktuellen Covid-19-Pandemie. Auch bei ABR seien alle Voraussetzungen einer Pandemie erfüllt: Resistente Erreger werden jeweils in unterschiedlicher Häufigkeit in den einzelnen Ländern übertragen, betroffene PatientInnen können schwer erkranken oder sterben und die Infektionserreger werden durch Reisen sowie internationalen Handel weltweit verbreitet. Deutschland habe diese Problematik erkannt und mit dem Infektionsschutzgesetz entsprechende Maßnahmen ergriffen. Aber es gelte, auch weltweit Verantwortung zu zeigen und beim Thema ABR stärker global zu agieren.
Im Fokus: Mensch
Im Folgenden gab es jeweils einen Fokus-Teil zum Bereich Mensch, Tier und Umwelt. ExpertInnen aus Human- und Tiermedizin, Gesundheitswissenschaften und Agrarwissenschaften stellten ihre Analysen und wissenschaftlichen Untersuchungen vor. Verschiedene Hotspots der Resistenzentwicklung wurden dabei unter die Lupe genommen. Jeder Fokus-Teil startete mit einer kurzen Theaterszene – Aufzeichnungen einer Aufführung der Straßentheatergruppe Schluck & weg aus dem vergangenen Jahr.
Im Fokus-Teil Mensch thematisierte der Kinderarzt Roland Tillmann die viel zu häufigen Antibiotika-Verschreibungen in Arztpraxen. Er berichtete über das Bielefelder Modellprojekt AnTiB, das durch die interaktive Entwicklung von Leitlinien auf lokaler Ebene die Verschreibungskultur nachhaltig verändert hat.
Jens Holst, Professor für Medizin mit Schwerpunkt Global Health am Fachbereich Pflege- und Gesundheit der Hochschule Fulda betrachtete Antibiotika-Resistenzen als systemische Herausforderung. Mit Medizintechnologie und Medikamenten allein wird man das Problem nicht in den Griff bekommen, so Holst. Notwendig sei „eine angemessene Beachtung aller gesundheitsrelevanten Politik- und Lebensbereiche wie Bildung, Arbeit, Wohnen, Ernährung, Verkehr, Umwelt, Sicherheit, Familie oder Freizeit“.
Im Fokus: Tier
Im Fokus-Teil Tier präsentierte Dr. Julia Steinhoff-Wagner die Ergebnisse einer Studie, die sie an der Landwirtschaftlichen Fakultät der Universität Bonn durchgeführt hat. Die Agrarwissenschaftlerin schilderte nicht nur eingehend die Problemlage im Bereich Nutztierhaltung. Sie stellte auch effektive Maßnahmen im Tiergesundheits-Management vor, um den Antibiotika-Einsatz in der Tierproduktion wirksam zu reduzieren. So hätten Reinigungs- und Desinfektionsmaßnahmen, eine veränderte Fütterung, aber auch geringere Tierdichte und kleinere Gruppen aus Sicht der Wissenschaft zwar positive Auswirkungen auf die Tiergesundheit. Aus Sicht der Veterinärämter würden diese Maßnahmen jedoch negativ bewertet – vor allem, weil es schwierig ist, eindeutige Nachweise für den Nutzen der Maßnahmen zu erbringen. Die Erhebung umfassender Daten ist für die Betriebe kaum praktikabel. Eine überbetriebliche Auswertung von Daten zum Gesundheits-Monitoring und Beratung der LandwirtInnen könne hier einen deutlichen
Mehrwert bieten. Letztlich müssten sich Nachhaltigkeitsinvestitionen und speziell Tierbeobachtungsleistungen für die LandwirtInnen aber auch lohnen.
Stig Tanzmann, Agrarexperte bei Brot für die Welt, skizzierte die aktuellen Problemfelder in den Bereichen Lebensmittelproduktion, Ernährung und globaler Handel. Er belegte mit aktuellen Zahlen, dass die deutsche Landwirtschaft vor allem bei der Fleischproduktion zu einem führenden Exporteur von Billigwaren geworden ist – Gemüse und Obst würden dagegen kaum noch auf deutschen Äckern produziert. Tanzmann plädierte für eine Wende in der Agrarpolitik und skizzierte Wege aus der Krise. Notwendig sei eine Abkehr von Intensivtierhaltung, Qualzucht und massivem Fleischkonsum.
Im Fokus: Umwelt
Der Fokus-Teil Umwelt begann mit einer Präsentation von Dennis Schmiege, Doktorand im Fachbereich Geografie der Universität Bonn. Schmiege hatte im Rahmen einer wissenschaftlichen Untersuchung am Institut für Urban Public Health der Uniklinik Essen das Vorkommen multiresistenter E. coli Erreger in häuslichen Abwässern untersucht. Das Ergebnis: Haushalte spielen eine wichtige Rolle beim Eintrag resistenter E. coli-Keime in das kommunale Abwasser. Besonders hoch ist die Konzentration resistenter Bakterien in den Wintermonaten. In den Wohngebieten gut situierter BürgerInnen in Stadtrandlage war die Keimbelastung außerdem weitaus niedriger als in sogenannten Brennpunktvierteln.
Prof. Martin Exner berichtete über Erkenntnisse aus dem Forschungsprojekt HyReKA, das Antibiotika-Resistenzen im Wasserkreislauf untersucht hat. Bis 2020 leitete er das Institut für Hygiene und öffentliche Gesundheit der Uniklinik Bonn. Exner hob insbesondere die Bedeutung des Abwasserreservoirs im Krankenhaus hervor: „Wir finden so besorgniserregende hohe Konzentrationen im Waschbecken, Dusch- und Toiletten-Abwasser, denen der Patient direkt exponiert ist, wie wir es in weiteren Bereichen des Abwasser und auch in Flüssen niemals finden. Die Ergebnisse des HyReKA Projektes waren eine wichtige Motivation zur Erstellung der Empfehlung der Kommission für Krankenhaushygiene und Infektionsprävention beim RKI, die 2020 erschien.“ Paul Kröfges, Gewässerschutzexperte des BUND, plädierte deshalb dafür, Kläranlagen, in die Klinikabwässer eingeleitet werden, mit einer zusätzlichen Reinigungsstufe auszustatten. Mit herkömmlicher Klärtechnik ließen sich weder antibiotische Rückstände noch multiresistente Keime effektiv herausfiltern.
Politische Forderungen und Strategien
Nach den drei Fokus-Teilen weitete Astrid Berner-Rodoreda, die am Heidelberger Institut für Global Health forscht, den Blick noch einmal für die globale Perspektive: „Wo bleibt das Globale?“ lautete der Titel Ihres Vortrags, der u.a. das fehlende Forschungsengagement der EU in den Blick nahm.
Abschließend wurden in mehreren Arbeitsgruppen Strategien entwickelt und Pläne für die weitere politische Arbeit geschmiedet. Die Zivilgesellschaft sei bisher beim Thema Antibiotika-Resistenzen zu wenig aktiv, hieß es in der Diskussion. Sie müsse dringend eine prominentere Rolle einnehmen, um die Probleme im Bereich Mensch, Tier und Umwelt gezielt in die Öffentlichkeit zu tragen. Die TeilnehmerInnen befürworteten unter anderem eine gemeinsame Aktion verschiedenster zivilgesellschaftlicher Organisationen zur Weltantibiotikawoche im November, ein Fachtreffen zur Planung soll im frühen Herbst stattfinden.
Besuchen Sie unsere Konferenz-Webseite!
Sämtliche Dokumente und Aufzeichnungen zu unserer Konferenz finden Sie unter https://bukopharma.de/konferenz/index.html. Unsere Konferenz-Webseite bietet sämtliche Vorträge als Video und auch als pdf-Dokument. Die Podiumsdiskussion vom 1. Mai können Sie sich hier ebenfalls noch einmal ansehen. Auch unser Forderungskatalog steht zum Download bereit. Ein Strategiepapier ist noch in Arbeit, wird aber ebenfalls zu einem späteren Zeitpunkt auf der Webseite zu finden sein. (CJ)
Bilder © Jörg Schaaber
Artikel aus dem Pharma-Brief 5/2021, S.4
Nutzenbewertung in Deutschland: Die Spreu vom Weizen trennen
Erfolgsmodell mit kleinen Schönheitsfehlern
Seit zehn Jahren wird bei allen neuen Medikamenten in Deutschland geprüft, ob sie PatientInnen tatsächlich besser helfen als die bislang verwendeten. Zeit für eine Bilanz.
Am 1. Januar 2011 trat das Arzneimittelmarktneuordnungsgesetz – kurz AMNOG – in Kraft. Hinter dem Wortungetüm steht ein klarer Auftrag: die systematische Prüfung des Nutzens aller Neueinführungen auf dem deutschen Arzneimittelmarkt. Das Gesetz verfolgt zwei Ziele: die Versorgung zu verbessern und die Krankenversicherung finanzierbar zu halten.
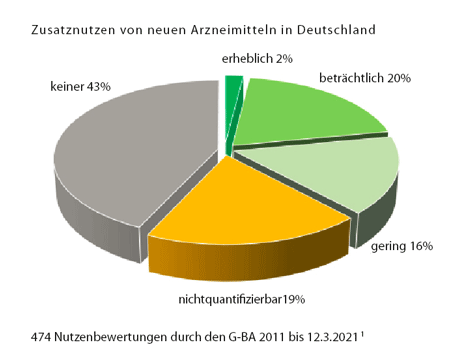
Die wichtigste Erkenntnis aus beinahe 500 Bewertungen (siehe Grafik auf S. 3):[1] Fast die Hälfte der neuen Medikamente (43%), die in den letzten zehn Jahren neu auf den Markt gekommen sind, bieten den PatientInnen keine Vorteile im Vergleich zu der bisher üblichen Behandlung. Gerade einmal 2% sind tatsächlich therapeutische Durchbrüche, immerhin 20% bringen beträchtliche Vorteile, aber 16% sind nur geringfügig besser. Problematisch ist der hohe Anteil der Medikamente mit unklarem Zusatznutzen (19% „nicht quantifizierbar“): Hier ist die Datenlage dermaßen unklar, dass man noch gerade erkennen kann, dass es vermutlich Vorteile gibt, aber sie unmöglich quantifizierbar sind.
All dies bietet jedoch ein zu rosiges Bild. Die genannten Zahlen beziehen sich auf die beste Bewertung eines Wirkstoffs. Häufig bieten neue Medikamente aber nicht allen Erkrankten Vorteile. Wenn die Urteile auf die unterschiedlichen PatientInnengruppen heruntergebrochen werden, findet sich bei rund 60% kein Zusatznutzen.
Dazu kommt: Medikamente gegen seltene Krankheiten (Orphan Drugs)[2] bekommen ohne Prüfung einen Zusatznutzen bescheinigt, solange sie die Krankenversicherung nicht mehr als 50 Millionen Euro pro Jahr kosten. Hier kann der Zusatznutzen also völlig fiktiv sein. Er ist gesetzlich vorgeschrieben, statt auf einer wissenschaftlichen Bewertung zu beruhen. Diese juristisch bedingte Bevorzugung betrifft jede fünfte Bewertung.[3]
Schlechte Studien
Vor allem bei den Waisenmedikamenten ist die Studienlage oft unbefriedigend. Die Zahl der untersuchten PatientInnen ist nicht selten unnötig klein oder die Dauer der Studien zu kurz, was sichere Aussagen über den Nutzen unmöglich macht. Es gibt oft keine Vergleiche gegen andere Behandlungsstrategien, obwohl sie vorhanden sind. Und mitunter wird gar nicht gemessen, was für die Betroffenen tatsächlich spürbare Verbesserungen bringt. Aufgrund dieser Unsicherheiten machen Waisenmedikamente den Löwenanteil der Medikamente mit nicht quantifizierbarem Zusatznutzen aus.
Allerdings wird auch bei anderen neuen Wirkstoffen allzu oft kein direkter Vergleich zu existierenden Medikamenten gemacht. Dadurch ist es fast unmöglich zu beurteilen, ob die Neuerung tatsächlich einen Vorteil bietet. Während sich die Hersteller zu Anfang des AMNOGs noch darauf berufen konnten, dass die Nutzenbewertung viel zu neu sei und die Zulassungsbehörden sich doch mit dem Vergleich gegen Placebo zufriedengegeben hätten, zieht dieses Argument heute nicht mehr – es wird von den Firmen aber trotzdem immer noch bemüht. Ein Trend zu mehr direkt vergleichenden Studien ist leider nicht zu erkennen. Insofern vergoss der Chef des Pharmaverbandes Vfa kürzlich Krokodilstränen, als er beklagte, dass fast die Hälfte der Studien vom G-BA für die Nutzenbewertung nicht berücksichtigt würde.[4] Das liegt nämlich einfach daran, dass sie dafür schlicht ungeeignet sind. Über die Jahre liegen für rund die Hälfte der neuen Medikamente keine direkten Vergleiche zum bisherigen Therapiestandard vor.[5]
Ein Problem sind auch die oft verfrühten Zulassungen bei noch gar nicht abgeschlossenen Studien. Solche Zwischenauswertungen lassen die Neuerungen manchmal in einem zu positivem Licht erscheinen. Zwei krasse Fälle betrafen zwei Krebsmedikamente, die vom G-BA einen Zusatznutzen zugesprochen bekamen. Später stellte sich heraus, dass der Wirkstoff Olaratumab unwirksam ist[6] und Ingenolmebutat Hautkrebs sogar häufiger macht.[7] Beide Mittel wurden verboten.
Her mit den Daten
Deutschland war mit der Einführung einer Nutzenbewertung spät dran. Andere europäische Staaten haben einen solchen Realitäts-Check schon seit vielen Jahren. Dafür bietet das deutsche Gesetz aber einen bedeutsamen Vorteil im Vergleich zu unseren Nachbarländern. Es verpflichtet nämlich den Anbieter, die vollständigen Studiendaten zu seinem Produkt zur Bewertung einzureichen, also auch den Teil, der gewöhnlich selbst für die Fachwelt unsichtbar bleibt.
Um sich ein Bild von solchen Geheimniskrämereien zu machen, ein Vergleich: Aus allen öffentlichen Quellen zusammengenommen[8] sind gerade einmal gut die Hälfte aller Studienergebnisse verfügbar, im AMNOG-Verfahren sind es dagegen nahezu 90%.[9] Das erlaubt eine viel fundiertere Einschätzung zum tatsächlichen Nutzen eines neuen Präparats.
Transparenz macht den Unterschied
Dazu ein Beispiel: Während das für England und Wales zuständige NICE dem Brustkrebsmedikament Palbociclib einen Zusatznutzen zusprach, konnte der G-BA keine Vorteile entdecken. Der Unterschied: Bei den Nachbarn lagen nur unvollständige Informationen zu dem Medikament vor. Der Hersteller behauptete, er hätte noch keine Daten dazu, ob die Frauen durch das neue Medikament tatsächlich länger überleben, aber bestimmte Messwerte gäben einen starken Hinweis dafür. Aufgrund der vollständigen Daten konnte in Deutschland demgegenüber errechnet werden, dass kein Überlebensvorteil durch Palbociclib vorhanden war. In England wurden die entscheidenden Angaben für die Bewertung vor der Veröffentlichung der Daten geschwärzt.[10] Es bleibt deshalb unklar, welche Zahlen dem NICE für die Entscheidung vorlagen. Die Begründung der Behörde bezog sich nicht auf einen Überlebensvorteil, sondern lediglich auf die besseren Messwerte, offensichtlich ein Trugschluss.
Verbesserungen angestoßen
In einem Punkt haben die Anbieter durch das Nutzenbewertungsverfahren auf jeden Fall dazugelernt. Während es zu Beginn des AMNOG-Verfahrens nur selten Daten zur Lebensqualität gab, legen heute drei Viertel der Anbieter auch Angaben vor, ob sich die PatientInnen durch die Behandlung mit dem neuen Präparat auch wirklich besser fühlen. Allerdings geschieht das oft noch halbherzig: Denn wenn die Lebensqualität nur bis zum Zeitpunkt einer Verschlechterung der Erkrankung gemessen wird (also z.B. zum Zeitpunkt, wo sich bei Krebs Metastasen bilden), fehlen wesentliche Informationen. Für die Erkrankten ist es aber sehr wichtig zu wissen, wie gut es ihnen in der verbleibenden Lebensspanne noch gehen mag.
Kräftig gespart
Finanziell hat die Nutzenbewertung die Krankenkassen und damit alle Versicherten deutlich entlastet. Zwar wurde erst nach acht Jahren, 2018, die vom Gesetzgeber angepeilte jährliche Ersparnis von zwei Milliarden Euro erreicht, für 2020 wurden aber durch die ausgehandelten Rabatte schon fast vier Milliarden Euro eingespart. Trotzdem bleiben die rasant steigenden Preise für neue Arzneimittel, die inzwischen bis in den Millionenbereich pro PatientIn reichen, ein Problem.
Was kommt in der Praxis an?
Ein Kritikpunkt am AMNOG-Verfahren ist, dass die detaillierten Erkenntnisse darüber, welche Medikamente für welche Zielgruppen Vorteile bringen, in der Krankenversorgung nicht ankommen. Ein Indiz dafür ist, dass auch neue Medikamente, die keinerlei Zusatznutzen haben, häufig verordnet werden. Damit sich das ändert, hat der Gesetzgeber verfügt, dass die Ergebnisse der Nutzenbewertung auf den Computern in Arztpraxen dargestellt werden müssen. Auch PatientInnen können sich selber schlau machen. Auf den Seiten des IQWiG gibt es verständliche Zusammenfassungen der Vor- und Nachteile von neuen Arzneimitteln.[11]
 Verbesserungen möglich
Verbesserungen möglich
Um die Preisspirale zu durchbrechen, gibt es mehrere Vorschläge. Der wichtigste ist vielleicht die rückwirkende Preisfestsetzung. Denn bislang beginnt der ausgehandelte Rabatt für ein neues Arzneimittel erst nach zwölf Monaten. Auf diese Weise können Anbieter – auch die, die ein Mittel ohne jeden Zusatznutzen auf den Markt bringen – ein Jahr lang kräftig absahnen.
Ein besonderes Problem sind die teuren Krebsmedikamente. Wenn die Vergleichstherapie schon 50.000 € oder mehr kostet, bedeutet ein neues Mittel für den Anbieter selbst dann ein gutes Geschäft, wenn wegen fehlenden Zusatznutzens kein höherer Preis erzielt werden kann.
Ein weiterer Vorschlag betrifft den oft enorm hohen Anfangspreis. Firmen sollen offenlegen, wie viel Geld sie in die Forschung gesteckt haben – und inwieweit ihr Produkt auch auf den Ergebnissen staatlich geförderter Forschung an Universitäten und öffentlichen Instituten beruht.
Auch die Nutzenbewertung sollte in einigen Punkten strenger werden. Der Freibrief für Waisenmedikamente unter 50 Millionen Euro Jahresumsatz muss entfallen, und Arzneimittel, die unter Auflagen zugelassen wurden, sollten immer nur eine befristete Bewertung erhalten.[12] Bei solchen Arzneimitteln kann der G-BA inzwischen vom Anbieter weitere anwendungsbegleitende Studien verlangen, um die Erkenntnislage zu verbessern. Bislang fehlt es aber an Sanktionsmöglichkeiten, wenn diese Auflage nicht erfüllt wird.
Undurchsichtige Expertise
Ein weiterer Schwachpunkt des Verfahrens bleibt voraussichtlich noch länger erhalten: Die Intransparenz bei den an Anhörungen beteiligten ExpertInnen. Sie stellen einen wichtigen Beeinflussungsfaktor bei der Nutzenbewertung dar. Während bei den IndustrievertreterInnen offensichtlich ist, welche Interessen sie vertreten, bleibt bei VertreterInnen von Fachgesellschaften die Verstrickung mit den Anbietern der diskutierten Arzneimitteln undurchsichtig – und nicht wenige pflegen enge Kontakte zu den Firmen. Es müssen bei den Anhörungen zwar detaillierte Erklärungen zu Interessenkonflikten vorgelegt werden, diese sind aber nur in absolut kondensierter Form auf der Website des G-BA einzusehen. So kann die Öffentlichkeit nicht mehr erkennen, dass Professor X zwar offiziell eine Fachgesellschaft vertritt, aber gleichzeitig dem Hersteller als Berater für genau das Produkt dient, über dessen Nutzen gerade diskutiert wird. Als Argument gegen eine vollständige Veröffentlichung wird der Schutz persönlicher Daten vorgebracht. Aber oft hat derselbe Professor gar kein Problem, (einen Teil seiner) Interessenkonflikte in internationalen Fachzeitschriften relativ detailliert anzugeben, weil diese sonst den Artikel schlicht nicht veröffentlichen würden.
Bei allen Fortschritten, die das AMNOG gebracht hat, gibt es also noch einigen Verbesserungsbedarf. (JS)
Eine kürzere Vorfassung dieses Artikels erschien in Gute Pillen – Schlechte Pillen (2021) Nr. 3, S. 18
Screenshot Nutzenbewertung ©Auszug aus dem Pharma-Brief
Grafik Zusatznutzen © www.g-ba.de/service/veranstaltungen/10-jahre-amnog
Artikel aus dem Pharma-Brief 3-4/2021, S.1
[1] G-BA-Tagung „10 Jahre AMNOG“ am 19.3.2021 www.g-ba.de/service/veranstaltungen/10-jahre-amnog
[2] Orphan Drugs (Waisenmedikamente) Arzneimittel gegen Krankheiten, von denen nicht mehr als 5 von 10.000 Menschen betroffen sind
[3] Bewertungen bis 31.12.2019. Storm A (Hrsg.) (2020) AMNOG-Report 2020. S. 215
[4] Han Steutel (Vfa) auf der G-BA-Tagung „10 Jahre AMNOG“ am 19.3.2021
[5] Thomas Kaiser (IQWiG) auf der G-BA-Tagung „10 Jahre AMNOG“ am 19.3.2021
[6] Pharma-Brief (2019) Zu früh ist unzuverlässig. Nr.1, S. 4
[7] Pharma-Brief (2020) Ungesunde Eile. Nr. 2, S. 6
[8] Veröffentlichung in Fachzeitschriften, Bericht der Zulassungsbehörde, Studienregister
[9] Untersucht anhand von 15 AMNOG-Verfahren. Köhler M. u.a. (2015) BMJ; 350, S. h796
[10] Pharma-Brief (2019) Die nächste Schlappe. Nr. 2, S. 6
[11] www.gesundheitsinformation.de/vom-iqwig-bewertete-wirkstoffe.html
[12] Für Orphan Drugs mit bis zu 50 Mio. € Umsatz führt der G-BA selbst eine Teilbewertung durch.
Molnupiravir: Öffentlich entdeckt – privat kassiert?
Covid-19 Medikament aus der Uni
Der Wirkstoff Molnupiravir könnte wichtig werden bei der Behandlung von Covid-19. Dass die Emory University den größten Teil dazu beigetragen hat, geht meist unter. Doch für die Diskussion um den Zugang spielt das eine bedeutende Rolle. Und der öffentliche Druck hat jetzt gewirkt: Der Medicines Patent Pool bekommt die Rechte für den Wirkstoff.
Noch ist nicht sicher, dass Molnupiravir die Erwartungen erfüllt, auch wenn die Presse begeistert darüber berichtet, dass der Wirkstoff die Sterblichkeit bei Covid-19 PatientInnen senken kann. Es liegen aber lediglich Daten vor, die der Anbieter MSD[1] auf einer Pressekonferenz nannte. Erst eine Publikation der Studie in einer Fachzeitschrift wird eine einigermaßen seriöse Einschätzung ermöglichen. Dann braucht es noch die Zulassung und eine Empfehlung der Weltgesundheitsorganisation. Für den Fall, dass sich der Nutzen erhärtet, ist es aber wichtig, dass frühzeitig die Voraussetzungen für eine ausreichende Produktion geschaffen werden.
Wer forscht denn da?
In den Medien wird oft nicht einmal erwähnt, dass der Emory University in den USA das Hauptverdienst für die Entdeckung von Molnupiravir zukommt und auch MSD erwähnt die Rolle von Emory eher am Rande. Seit 2013 forscht die Uni an dem Wirkstoff gegen verschiedene Viren. Dabei zeigte sich, dass er im Tierversuch gegen Influenza, aber auch gegen das Coronavirus MERS hilft. Zu Beginn der Corona-Pandemie im März 2020 entschied sich Emory, Molnupiravir an eine Pharmafirma zu lizensieren. Denn der Uni fehlte es an ausreichenden Kapazitäten zur schnellen Durchführung von klinischen Studien. Der Lizenznehmer Ridgeback Biotherapeutics ging zwei Monate später eine Kooperation mit dem Pharmamulti MSD ein. Die Studie, über die jetzt breit berichtet wird, begann im Juni diesen Jahres[2] und die Rekrutierung weiterer PatientInnen wurde nach einer geplanten Zwischenauswertung im Oktober vorzeitig gestoppt.[3]
Die Emory University hatte dagegen sechs Jahre lang mit Molnupiravir und Vorgängersubstanzen umfangreiche vorklinische Studien gegen eine Reihe von Krankheitserregern durchgeführt und dafür staatliche Zuschüsse von schätzungsweise 35 Mio. US$ erhalten. Die Universität hat vier Patentanträge für den Wirkstoff eingereicht, darunter einen spezifisch als Arzneimittel gegen SARS-CoV-2. Wegen der öffentlichen Förderung besitzt auch die US-Regierung ein Zugriffsrecht auf Molnupiravir.[4] Das wurde aber offensichtlich nicht genutzt. Zwar haben sich die USA bereits 1,7 Millionen Therapieeinheiten des Medikaments gesichert, allerdings zu dem stolzen Preis von 712 US$ pro PatientIn. Das macht in der Summe rund 1,2 Milliarden US$. Der Vertrag enthält eine Option auf weitere Dosen für bis zu 2,5 Mrd. US$. Nur für den Fall, dass MSD das Produkt nicht mehr herstellt, hat sich die US-Regierung alle Rechte an Molnupiravir gesichert.[5] Das Vereinigte Königreich hat sich ebenfalls bereits Kontingente für eine halbe Million PatientInnen reserviert, der Preis ist nicht öffentlich bekannt.[6]
Enorme Gewinne
Melissa Barber (Harvard University) und Dzintars Gotham (Kings College, London) schauten sich die realen Lieferpreise für den reinen Wirkstoff (API) an.[7] Der durchschnittliche Kilopreis betrug 2.162 US$. Dabei handelte es sich um kleine Liefermengen von 1-20 kg. Ahlqvist und KollegInnen kalkulierten den Kilopreis für die API in der regulären Produktion mit 799 US$ und bei Recycling der notwendigen Lösungsmittel mit lediglich 427 US$.[8]
Barber und Gotham errechneten auf Basis der gegenwärtigen Lieferpreise Kosten von rund 20 US$ pro PatientIn – einschließlich 10% Gewinn und Steuern.[9] Damit stünden den von der US-Regierung mit MSD vereinbarten 1,2 Mrd. US$, gegenwärtig Produktionskosten von 34 Mio. US$ gegenüber, bei Massenproduktion und optimierten Prozessen wären es gerade einmal 7,4 Mio. US$. Wahrhaftig eine gewaltige Gewinnspanne.
Zugang – es tut sich was
Und wie sieht es mit dem Rest der Welt aus? Viel besser als bei den Covid-19 Impfstoffen, wo nur Oxford University und AstraZeneca einer indischen Firma frühzeitig eine Produktion für ärmere Länder gestattet hatten und bedeutende Impfstoffmengen in diesen Staaten auch angekommen sind.[10]
Zunächst hatte MSD acht indischen Generikafirmen Lizenzen für die Herstellung von Molnupiravir für Länder mit mittlerem und niedrigen Einkommen erteilt. Die genauen Bedingungen sind unbekannt.[11] MSD teilte lediglich mit, dass „die relativen Möglichkeiten der Länder, ihre Antwort auf die Pandemie zu finanzieren,“ berücksichtigt würden.[12]
Durchbruch für Pool
Ende Oktober kam dann vom Medicines Patent Pool (MPP) die Erfolgsmeldung: Molnupiravir ist das erste Covid-19-Medikament, für das der MPP die Lizenzrechte erhält, einschließlich des Zugriffs auf die Daten aus klinischen Tests. Damit können Generikafirmen den Wirkstoff unkompliziert und preiswert herstellen. Und das Beste: Solange die Pandemie nicht zu Ende ist, verzichtet MSD auf die übliche Lizenzgebühr. Danach beträgt sie 5% des Verkaufspreises für den öffentlichen Sektor und 10% für den privaten. Einziger Wermutstropfen: Zwar können die Medikamente in 105 Ländern genutzt werden,[13] aber stark von Covid-19 betroffene Länder mittleren Einkommens wie Brasilien, Argentinien und Peru gehören nicht dazu.[14]
Eigentlich war der MPP von der WHO von Anfang an dafür vorgesehen, günstige Lizenzen für Covid-19-Produkte einzusammeln. Doch bis jetzt blieb der Pool leer, weil die Hersteller den MPP boykottierten. Das fiel ihnen leicht, weil wichtige Industrieländer – darunter auch Deutschland – dem MPP ebenso die Unterstützung verweigerten wie einer temporären Aufhebung der Patentrechte durch die WTO (Waiver). Der Schutz der einheimischen Konzerne ist den reichen Ländern offensichtlich immer noch wichtiger, als die globale Versorgung mit Medikamenten und Impfstoffen. Doch der öffentliche Druck für den globalen Zugang nimmt massiv zu. Zuletzt haben sich auch hunderte WissenschaftlerInnen und hochrangige PolitikerInnen den Protesten angeschlossen. Es sieht so aus, als könne die Stimmung kippen. Angesichts von Tausenden durch Impfung und Behandlung vermeidbaren Todesfällen, ist das überfällig. (JS)
Artikel aus dem Pharma-Brief 8-9/2021, S. 4
[1] In den USA und Kanada Merck Sharp & Dohme Corp. genannt
[2] https://clinicaltrials.gov/ct2/show/record/NCT04939428 [Zugriff 20.10.2021]
[3] www.merck.com/news/merck-and-ridgebacks-investigational-oral-antiviral-molnupiravir-reduced-the-risk-of-hospitalizationor-death-by-approximately-50-percent-compared-to-placebo-for-patients-with-mild-or-moderat [Zugriff 20.10.2021]
[4] Abinader LG (2021) US government rights in patents on Molnupiravir, based upon funding of R&D at Emory University. KEI www.keionline.org/36648 [Zugriff 20.10.2021]
[5] Ardizzone K (2021) U.S. Government’s $1.2 Billion Contract for Merck’s Investigational COVID-19 Drug Molnupiravir Redacts IP Terms, Contains Donation Clause and Very Limited Technology Transfer License. KEI www.keionline.org/36698 [Zugriff 20.10.2021]
[6] Walker P and Sample I (2021) No 10 to buy new antiviral treatments for Covid in time for winter. Guardian 20 Oct. www.theguardian.com/world/2021/oct/20/no-10-to-buy-new-antiviral-treatments-for-covid-in-time-for-winter
[7] Lieferungen von Mai bis Juli 2021
[8] Ahlqvist GP et al. (2021) Progress Toward a Large-Scale Synthesis of Molnupiravir (MK-4482, EIDD-2801) from Cytidine. ACS Omega; 6, p 10396 https://doi.org/10.1021/acsomega.1c00772
[9] Barber MJ and Gotham D (2021) Estimated cost-based generic prices for molnupiravir for the treatment of COVID-19 infection. https://scholar.harvard.edu/melissabarber/publications/estimated-cost-based-generic-prices-molnupiravir-treatment-covid-19
[10] Anteil der tatsächlichen Lieferungen an Länder mit niedrigem und niedrigem mittleren Einkommen insgesamt (Direktverkäufe, Spenden und Covax. Stand 12.10.2021): Oxford/AstraZeneca 70,7%, Johnson&Johnson 30,1%, Moderna 12,8%, Biontech 5,9%. Quelle: Malpani R and Maitland A (2021) Dose of Reality: How rich countries and pharmaceutical corporations are breaking their vaccine promises. The People’s Vaccine. 20 Oct., p 25 www.oxfam.de/system/files/documents/a_dose_of_reality-briefing_note.pdf
[11] Guarascio F and Erman M (2021) Merck COVID-19 pill sparks calls for access for lower income countries. Reuters 17 Oct. www.reuters.com/business/healthcare-pharmaceuticals/merck-covid-19-pill-sparks-calls-access-lower-income-countries-2021-10-17
[12] MSD (2021) Merck Announces Supply Agreement with U.S. Government for Molnupiravir, an Investigational Oral Antiviral Candidate for Treatment of Mild to Moderate COVID-19. Press release 9 June www.merck.com/news/merck-announces-supply-agreement-with-u-s-government-for-molnupiravir-an-investigational-oral-antiviral-candidate-for-treatment-of-mild-to-moderate-covid-19/
[13] https://medicinespatentpool.org/licence-post/molnupiravir-mol#country-list0 [Zugriff 4.11.2021]
[14] Lei Ravelo J (2021) Merck expands manufacturing for molnupiravir. But questions remain. DEVEX, 28 Oct. www.devex.com/news/merck-expands-manufacturing-for-molnupiravir-but-questions-remain-101914 [Zugriff 4.11.2021]
Medikamente: Essenzieller Mangel
Neue WHO-Liste unentbehrlicher Arzneimittel
Am 1. Oktober wurde die 22. Ausgabe der Essential Medicines List (EML) veröffentlicht.[1] Einen besonderen Augenmerk legt die Weltgesundheitsorganisation (WHO) darin auf nicht-übertragbare Erkrankungen (NCDs). Bemerkenswert: Die begleitende Forderung nach einer beratenden Arbeitsgruppe zu finanziellen Zugangshürden.
Alle zwei Jahre wird die Modellliste unentbehrlicher Arzneimittel aktualisiert. Seit 1977 führt sie die Präparate, die weltweit in Gesundheitssystemen jederzeit in adäquater Menge, guter Qualität und zu einem erschwinglichen Preis verfügbar sein sollten. Bereits zu Beginn der jüngsten Überarbeitung wurde jedoch eine Konfliktlinie deutlich, die bereits in der Vergangenheit Thema war.
Zwar waren zuletzt mehrere, selbst für reichere Länder finanziell schwer zu stemmende Präparate auf die Liste geholt worden, etwa gegen Hepatitis C.[2] Zivilgesellschaftliche Akteure kritisierten gleichwohl, dass das nicht ausreicht. So wurde die Forderung nach einer Liste lauter, die spezifisch „medizinisch unentbehrliche aber schwer finanzbare Arzneimittel“ benennt.
Knowledge Ecology International (KEI) argumentierte etwa: „Die EML spielt heute in Debatten um Zugang zu Medikamenten oftmals eine negative Rolle. Die geringe Anzahl patentierter Medikamente auf der EML wird regelmäßig als Beleg dafür herangezogen, dass Patente keine Barriere für den globalen Zugang zu unentbehrlichen Medikamenten sind.“[3] Nun geht die WHO in einem begleitenden Bericht zur neuen EML einen Schritt auf ihre KritikerInnen zu.
Theoretisch unentbehrlich, praktisch unbezahlbar
Während des Auswahlprozesses, so konstatiert das EML-ExpertInnenkomitee, habe man den Trend der kontinuierlichen Preissteigerungen bei neuen Präparaten zur Kenntnis genommen – besonders bei Krebs, Autoimmunerkrankungen, Infektionskrankheiten und seltenen Erkrankungen.[4] Nur wenige dieser Präparate hätten so bedeutende Vorteile, dass sie trotz des hohen Preises aufgenommen werden könnten, bei anderen sei der Zusatznutzen nicht groß genug und sie würden deshalb keinen Eingang in die Liste finden.
Das Komitee stellt ergänzend dazu fest, dass sich das Problem der Bezahlbarkeit auch bei älteren wichtigen Medikamenten zeige, zum Beispiel bei Insulin. Es sei eine anhaltende Herausforderung, solche Präparate verfügbar zu machen: „Für Länder niedrigen und mittleren Einkommens ist dies besonders wichtig, denn die Zahl der Menschen mit Erkrankungen, die solche Medikamente benötigen, nimmt stetig zu.“[3]
Der Report empfiehlt die Einrichtung einer permanenten EML-Arbeitsgruppe. Sie soll das Expertenkomitee unterstützen und so der WHO helfen, Instrumentarien für den verbesserten Zugang zu teuren Medikamenten zu entwickeln. Wie notwendig das ist, verdeutlicht symptomatisch das Therapiefeld Krebs.
Milliardengewinne statt breiter Versorgung
Für die EML 2021 lagen Vorschläge für 40 neue Wirkstoffe sowie für 16 neue Indikationen vor.[5] Diese betrafen neben NCDs auch Arbeitsfelder wie sexuelle und reproduktive Gesundheit, Tuberkulose und den Themenbereich Antibiotikaresistenzen. Allein 16 Vorschläge für Wirkstoffe und für 6 neue Indikationen gab es für den Bereich Krebs. Bereits bei der letzten Überarbeitung 2019 hatte die WHO 10 neue Krebspräparate in die Liste aufgenommen – seither fanden sich 56 darauf (plus 3 alternative Wirkstoffe). Neu hinzugekommen sind nun:
- Enzalutamid, als Alternative zu Abirateron bei Prostatakrebs,
- Everolimus, zum Einsatz bei speziellen Hirntumoren bei Kindern,
- Ibrutinib, bei chronischer lymphatischer Leukämie,
- sowie Rasburicase, zum Management des sogenannten Tumorlyse-Syndroms.
Symptomatisch für das von der WHO beklagte Finanzierungsproblem sind hingegen die Kandidaten, die ausdrücklich aus Kostengründen nicht aufgenommen wurden. Sogenannte Checkpoint-Inhibitoren etwa fanden keinen Eingang in die EML 2021, darunter Pembrolizumab, das von Merck als „Blockbuster“ Keytruda® vertrieben wird und allein 2020 einen Umsatz von über 14 Milliarden US-Dollar erzielte.[6] Die WHO verweist in diesem Kontext konkret auf die sehr hohen Preise, mit denen eine Überbelastung von Versorgungssystemen mit geringen Ressourcen einherginge. Verschärft würde dies durch die Notwendigkeit adäquater Diagnostik, um PatientInnen zu identifizieren, die von einer Behandlung profitieren könnten, die Unklarheit über die optimale Behandlungsdauer sowie die hohe Zahl an möglichen PatientInnen.
Schlüsselrolle für Biosimilars
Die große Aufmerksamkeit für das Thema Krebs kommt, obwohl auch bei anderen Erkrankungen die Finanzierung gravierende Probleme macht, nicht von ungefähr. Immer mehr Studien offenbaren die enormen Versorgungslücken weltweit. So wertete eine Studie in der Fachzeitschrift Lancet Oncology kürzlich Daten zum Zugang zu wichtigen Krebsmedikamenten in 82 Ländern aus, ein Großteil von ihnen auf der EML. Das Fazit war niederschmetternd: Selbst ältere Präparate wie Cisplatin oder Tamoxifen bedeuteten schwere finanzielle Bürden für die PatientInnen, viele neuere waren gar nicht erst verfügbar.[7]
Ein Faktor, den die WHO in jüngster Zeit stärker ins Rampenlicht gerückt hat, ist der Ausbau der Biosimilar-Produktion, bei Diabetes, als auch bei Krebs. Dabei gibt es hohe Hürden, wie kürzlich ein Beitrag der Süddeutschen Zeitung am Beispiel Indiens herausarbeitete und für den die Pharma-Kampagne Informationen lieferte.[8] Die regulatorischen Anforderungen sind so groß, dass sich die Entwicklung von Biosimilars für kleinere Firmen kaum lohnt. Entsprechend können große Konzerne ihre problematische Marktmacht sichern.
Es geht aber auch konkret um die Lizenzierung patentierter Wirkstoffe. Der EML Report weist dem Medicines Patent Pool (MPP) dabei eine zentrale Rolle zu. Er solle unter anderem die Möglichkeit von Lizenzen für Enzalutamid und Ibrutinib prüfen. So begrüßenswert die Unterstützung für das Engagement des MPP bei Krebs auch ist, wird sie allerdings eine grundlegende Krux nur schwerlich auflösen können, wie die an der Erstellung der 22. WHO-Liste beteiligte Expertin Ellen ‘t Hoen jüngst bilanzierte: „Ich vermute, dass es wesentlich schwieriger werden wird, Lizenzen für Krebsmedikamente zu bekommen als für HIV-Therapien, weil es für die Firmen so hochprofitable Produkte sind.“ [9] Nicht zuletzt deshalb steht zu erwarten, dass im globalen Süden auch Zwangslizenzen, bei Krebspräparaten schon von Ländern wie Thailand erfolgreich erprobt, verstärkt in Betracht gezogen werden. (MK)
Preiserhöhung ab 2022
Sechs Jahre haben wir die Abopreise stabil gehalten. Jetzt ist leider eine Erhöhung notwendig. Ab 2022 kostet das Einzelabo 26 € (statt 22 €) und das Institutionen- oder Auslandsabo 50 € (statt 42 €).
Die Kündigungsfrist beträgt vier Wochen zum Jahresende. Wir hoffen aber, dass Ihnen der Pharma-Brief seinen Preis wert ist und wir Sie weiterhin zu unseren LeserInnen zählen können. Kleiner Bonus: Wir arbeiten auch an einem neuen Layout, so dass das Heft noch lesbarer wird.
Umstrittene Entscheidungen
Das Grippemittel Oseltamivir wurde wieder nicht gestrichen. Dabei hatte es das Mittel nur wegen lückenhafter Studiendaten in die WHO-Liste geschafft. Seit einigen Jahren gilt als gesichert, dass es im Vergleich zu Placebo lediglich die Krankheitssymptome um einen Tag verkürzen kann, aber keine Leben rettet. Auch die Aufnahme von Insulinanaloga ist fragwürdig. Denn sie bieten keine nennenswerten Vorteile, sind aber teurer als Humaninsulin. Zivilgesellschaftliche Akteure hatten bei der EML 2019 die Listung der Analoga noch verhindern können. Die WHO rechtfertigt sich jetzt mit einer veränderte Ausgangslage bei Produktion und Preisen.[10]
Artikel aus dem Pharma-Brief 8-9/2021, S. 1
Bild © Minette Lontsie
[1] Außerdem wurde zeitgleich die 8. Ausgabe einer speziellen Liste für Kindermedikamente veröffentlicht.
[2] Pharma-Brief (2017) Weniger Medikamente – bessere Behandlung. 40 Jahre Liste unentbehrlicher Arzneimittel der WHO. Spezial Nr. 2
[3] Fletcher ER (2021) Forty new medicines & 16 new indications under consideration for WHO Esssential Medicines List. https://healthpolicy-watch.news/who-considering-40-new-medicines-16-new-indications-for-global-essential-medicineslist/#print [Zugriff 19.10.2021]
[4] WHO (2021) Executive summary. The Selection and Use of Essential Medicines 2021 Report of the 23rd WHO Expert Committee on the Selection and Use of Essential Medicines.
[5] WHO (2021) WHO Expert Committee on the Selection and Use of Essential Medicines. www.who.int/groups/expert-committee-on-selection-and-use-of-essential-medicines/23rd-expert-committee [Zugriff 19.10.2021]
[6] Merck (2021) Merck announces fourth-quarter and full-year 2020 financial results. www.merck.com/news/merck-announces-fourth-quarter-and-full-year-2020-financial-results/ [Zugriff 25.10.2021]
[7] Fundytus A et al. (2021) Access to cancer medicines deemed essential by oncologists in 82 countries: an international, cross-sectional survey. Lancet Oncology; https://doi.org/10.1016/ S1470-2045(21)00463-0
[8] Viciano A (2021) Wenn Überleben eine Frage des Geldes ist. Süddeutsche Zeitung. 6. August https://sz.de/1.5374274 [Zugriff 25.10.2021]
[9] Pharma Brief (2021) Unbezahlbar krank? Spezial Nr. 1
[10] Santos R (2021) New WHO Essential Medicines List Includes Controversial Insulin Analogues; Recommends Action on High Medicines Prices. Health Policy Watch 1 Oct. https://healthpolicy-watch.org/who-essential-medicines-insulin-analogues
Jetzt reinhören: Arzneimittel im globalen Süden
Neue Podcast-Reihe mit Stimmen aus Afrika und Mexiko
In den letzten Wochen wurden mehrere Akteure aus dem globalen Süden im Rahmen des Bildungsprojektes „Großbaustelle Arzneimittelversorgung“ interviewt. Die BUKO Pharma-Kampagne sprach mit AkteurInnen aus Mexiko, Sierra Leone und Malawi sowie anderen afrikanischen Ländern über den Zugang zu Gesundheitsleistungen, Arzneimitteln und medizinischem Equipment.
In fünf Podcast-Folgen kommen die InterviewpartnerInnen zu Wort. Sie berichten anhand spannender Geschichten, welche Baustellen im globalen Süden existieren, wenn eine gerechte und rationale Arzneimittelversorgung in den Blick genommen wird, wie sie mit ihnen umgehen und welche Werkzeuge für kommende Bauarbeiten auf den Baustellen benötigt werden.
Zu Gast ist zum Beispiel Rajab Lawe aus Malawi. Er gibt Einblicke in seine Arbeit für action medeor. Der gelernte Pharmazeut ist für die Qualitätssicherung zuständig und kontrolliert akribisch bei jeder Medikamenten-Lieferung, ob alles in Ordnung ist und sorgt dafür, dass es auch dabei bleibt: „Wir haben in unseren Lagerhäusern hier in Malawi Temperaturkontrollsysteme. Wir verwenden einen Wifi-Logger, mit dem wir die Temperatur alle 15 Minuten in Echtzeit überwachen können. Es gibt also Alarme, die uns normalerweise benachrichtigen, sobald es eine Temperaturabweichung gibt.“
Susanne Schröder von den Driving Doctors in Sierra Leone berichtet, wie sie die Mutter-Kind-Gesundheit mit ihrem mobilen Versorgungsangebot verbessern. Auf unserer Seite stehen die Podcast-Folgen zur Verfügung – Reinhören lohnt sich! (CK)
Artikel aus dem Pharma-Brief 8-9/2021, S. 3
Bild © Monstera, Bearbeitung Pharma-Brief
Gesundheit im Zeichen globaler Ungleichheit
Memento Preisverleihung 2021 mit Dr. Ayoade Olatunbosun-Alakija
Gleich drei Memento Preise wurden am 2. Dezember in einer digitalen Zeremonie vergeben. In der Veranstaltung wurden die dramatischen Herausforderungen deutlich, mit denen die Weltgemeinschaft konfrontiert ist – und auch die große Verantwortung Deutschlands. Die Keynote für den Abend kam live aus Nigeria.

Die Rede von Dr. Ayoade Olatunbosun-Alakija, Ko-Vorsitzende der African Vaccine Delivery Alliance der Afrikanischen Union, entfaltete eine enorme Wucht. Sie legte pointiert die krasse Ungerechtigkeit in der Covid-19-Versorgung weltweit offen und die fehlende Bereitschaft, notwendige strukturelle Änderungen zur Verbesserung der Lage anzugehen: „Wir lassen Menschen sterben aufgrund von Egoismus. Wir lassen Menschen sterben aufgrund von Gier.“
Insofern passte die Preisverleihung in der Kategorie „politischer Wille“ an Ottmar von Holtz (MdB 2017-2021) ins Bild. Zählte von Holtz in der vergangenen Legislaturperiode doch zu den wenigen Stimmen im Bundestag, die beharrlich die Bedeutung der nachhaltigen Entwicklungsziele für eine gerechte globale Gesundheitsversorgung in den Fokus gerückt haben. Er mahnte die Notwendigkeit schneller und dringend notwendiger Covid-19-Maßnahmen weltweit an, etwa auch in Form eines Patent Waivers bei der WTO. Ein Engagement, das Manuel Koch von der DAHW im Namen des Memento-Bündnisses würdigte.
Auch abseits der Pandemie – und gerade, weil sie droht, alles andere in den Schatten zu stellen – gilt es, auf andere vernachlässigte Gesundheitsbedürfnisse aufmerksam zu machen, so Nicola Kuhrt, Leiterin der Memento Medienjury. Deshalb zeichnete sie Olivia Kortas (Text) und Johannes De Bruycker (Fotos) mit dem Medienpreis in Form eines Recherchestipendiums aus. Ihr Konzept für eine Reportage zu gesundheitsgefährdenden Schimmelpilzgiften in Lebensmitteln in ärmeren Ländern wusste zu überzeugen.
Mit ihrer langjährigen Arbeit in der Tuberkulose-Bekämpfung konnte Claudia Denkinger bei der Forschungsjury punkten. Prof. Dr. August Stich, Chefarzt der Tropenmedizin am Klinikum Würzburg Mitte und Leiter der Jury, würdigte entsprechend ihr Engagement für eine bessere Tuberkulosediagnostik für ressourcenarme Settings.
In einer spannenden Diskussion wurde versucht, die Arbeit der Preistragenden zu verknüpfen. So wurde das Versäumnis der Politik kritisiert, in der Forschungsförderung nicht stärker auf konkrete Zugangsregelungen für die Endprodukte zu setzen. Die Folgen werden gerade bei Covid-19-Impfstoffen schmerzhaft deutlich. Abgerundet wurde der fruchtbare Austausch mit einigen Wünschen der Preistragenden an die neue Bundesregierung. Wichtigster Punkt: Eine pharmakritischere Haltung.
Max Klein von der BUKO Pharma-Kampagne, der die Veranstaltung moderierte, schloss die Preisverleihung mit einem Blick in die Zukunft ab: „Wir haben heute gehört, dass die Verantwortung Deutschlands für eine gerechte Gestaltung globaler Gesundheit groß ist. Wir haben zudem festgestellt: Der Rest der Welt nimmt sehr genau davon Kenntnis, ob Worten auch Taten folgen.“
Die Aufzeichnung der Preisverleihung finden Sie auf der Memento Homepage.
Infos gibt es auch auf Twitter: @memento_preis
Der Forschungspreis
 Claudia Denkinger vom Uniklinikum Heidelberg erforscht unter anderem, wie die Diagnostik bei der Tuberkulose-Bekämpfung im Globalen Süden vereinfacht werden kann. Bei ihrer Arbeit in ressourcenschwachen Ländern bedeutete eine fehlende Diagnose oft, dass sie die vermutete Erkrankung nicht behandeln durfte: „Und so habe ich viele Menschen unnötig sterben sehen.“ Mit 1,4 Millionen Todesfällen im vergangenen Jahr ist die heilbare Infektionskrankheit immer noch eine der häufigsten vernachlässigten Krankheiten weltweit. Sie entwickelt erfolgreich bessere Tests für Menschen, die mit HIV leben und durch Tuberkulose besonders gefährdet sind. Denn nur eine frühe Diagnose sichert ihnen gute Behandlungschancen.
Claudia Denkinger vom Uniklinikum Heidelberg erforscht unter anderem, wie die Diagnostik bei der Tuberkulose-Bekämpfung im Globalen Süden vereinfacht werden kann. Bei ihrer Arbeit in ressourcenschwachen Ländern bedeutete eine fehlende Diagnose oft, dass sie die vermutete Erkrankung nicht behandeln durfte: „Und so habe ich viele Menschen unnötig sterben sehen.“ Mit 1,4 Millionen Todesfällen im vergangenen Jahr ist die heilbare Infektionskrankheit immer noch eine der häufigsten vernachlässigten Krankheiten weltweit. Sie entwickelt erfolgreich bessere Tests für Menschen, die mit HIV leben und durch Tuberkulose besonders gefährdet sind. Denn nur eine frühe Diagnose sichert ihnen gute Behandlungschancen.
Der Medienpreis
Aflatoxine, gefährliche Giftstoffe, die von sich immer stärkerer verbreitenden Schimmelpilzen produziert werden, schwächen die Ärmsten und können sie ihr Leben kosten. Olivia Kortas und Johannes De Bruycker reisen mit ihrem Recherchestipendium nach Kenia, wo sich die Giftstoffe unter anderem in getrocknetem Fisch, Milchprodukten, Reis oder Mais, dem Grundnahrungsmittel, verstecken. „Die Aufnahme von geringen Mengen über einen langen Zeitraum kann mit Leberkrebs enden“, wissen die freien JournalistInnen schon jetzt. Aufgrund des Klimawandels verschärfen sich die Probleme. Produzieren werden sie eine Magazin-Reportage, um die kaum sichtbare Gesundheitskrise greifbar zu machen.
Der Politikpreis
 Für seinen Einsatz für eine gerechtere Gesundheitsversorgung weltweit wurde der ehemalige Grünen-Bundestagsabgeordnete Ottmar von Holtz mit dem Politikpreis ausgezeichnet. Er hat in der vergangenen Legislatur immer wieder die Perspektive des Globalen Südens eingenommen und anhand der Corona-Pandemie auf strukturelle Missstände in der Globalen Gesundheitsversorgung hingewiesen. In der Diskussion um schnelle und notwendige Covid19-Maßnahmen weltweit hat er immer wieder die nachhaltigen Entwicklungsziele in den Fokus gerückt und somit den Forderungen des Memento-Bündnisses nach einem nachhaltigen Aufbau von Gesundheitsstrukturen und sozialen Sicherungssystemen für alle Menschen weltweit Nachdruck verliehen.
Für seinen Einsatz für eine gerechtere Gesundheitsversorgung weltweit wurde der ehemalige Grünen-Bundestagsabgeordnete Ottmar von Holtz mit dem Politikpreis ausgezeichnet. Er hat in der vergangenen Legislatur immer wieder die Perspektive des Globalen Südens eingenommen und anhand der Corona-Pandemie auf strukturelle Missstände in der Globalen Gesundheitsversorgung hingewiesen. In der Diskussion um schnelle und notwendige Covid19-Maßnahmen weltweit hat er immer wieder die nachhaltigen Entwicklungsziele in den Fokus gerückt und somit den Forderungen des Memento-Bündnisses nach einem nachhaltigen Aufbau von Gesundheitsstrukturen und sozialen Sicherungssystemen für alle Menschen weltweit Nachdruck verliehen.
(MK/CK)
Artikel aus dem Pharma-Brief 10/2021, S.6
Bild Preisobjekt © Ansgar Silies
Bild Dr. Ayoade Olatunbosun-Alakija wurde uns zur freien Verfügung gestellt.
Bild Forschungspreis Claudia Denkinger © Tamara von Rechenberg
Bild Medienpreis Johannes De Bruycker und Olivia Kortas © Kasper Goethals
Bild Politikpreis Ottmar von Holtz © Kaminski
Europäische Nutzenbewertung kommt
Verbesserungen und viele kritische Punkte
Bereits seit mehreren Jahren gibt es Bestrebungen, die Bewertung neuer Arzneimittel in der EU zu vereinheitlichen und damit nationale Verfahren abzuschaffen. Erste Vorschläge stießen auf scharfe Kritik (wir berichteten[1],[2],[3]). Eine Einigung war lange nicht in Sicht. Im Windschatten von Covid-19 bahnt sich – von der Öffentlichkeit weitgehend unbemerkt – ein Ende der Geschichte an. Ein zwischen Mitgliedsstaaten, Kommission und Parlament abgestimmter Entwurf für eine europäische Nutzenbewertung liegt jetzt vor.[4] Wir berichten über Schwachstellen und Fortschritte im Sinne der PatientInnen.
Entscheidender Treiber bei der Vereinheitlichung der Nutzenbewertung in der EU war die Pharmaindustrie. Sie wollte sich nicht länger mit den verschiedenen Verfahren in den Mitgliedsstaaten auseinandersetzen, die unterschiedliche Anforderungen an die einzureichenden Unterlagen haben und teilweise zu verschiedenen Bewertungen kommen.
Aus Sicht der Hersteller ist es außerdem ein Ärgernis, dass bei vielen neuen Medikamenten kein Mehrnutzen für die PatientInnen erkannt wird und diese Mittel dann in etlichen Ländern von den Krankenkassen nicht erstattet werden.[5] Deshalb ging es der Industrie nicht nur um Vereinheitlichung, sie versuchte gleichzeitig eine Senkung der wissenschaftlichen Anforderungen durchzusetzen.
Die Kritik an den ersten Vorschlägen ist nicht ohne Folgen geblieben. Im aktuellen Gesetzentwurf der EU[6] spiegeln sich echte Fortschritte wider. Allerdings bleibt manches fragwürdig: Die Kommission räumt sich selbst weitgehende Rechte im Prozess der Bewertung ein und Hersteller erhalten Eingriffsmöglichkeiten, die inakzeptabel sind. Eine erhebliche Unsicherheit bleibt, weil etliche Bestimmungen im Detail erst nachträglich durch sogenannte delegierte Rechtsakte (Delegated Acts) von der Kommission selbst festgelegt werden sollen. Das öffnet die Tür für weitere Beeinflussungsversuche.
Langer Vorlauf nötig
Die europäische Nutzenbewertung soll drei Jahre nach ihrer Verabschiedung in Kraft treten, um dem Aufbau der notwendigen Strukturen und einer anschließenden Pilotphase genügend Zeit zu lassen. In drei Jahren soll die neue Behörde dann zuerst alle Krebsmedikamente und sogenannte neuartige Therapien (ATM) bewerten (erstere machen derzeit rund ein Drittel aller Neuzulassungen aus). Nach weiteren drei Jahren sollen Medikamente gegen seltene Erkrankungen folgen, alle übrigen Arzneimittel kommen noch zwei Jahre später an die Reihe.
Auch Medizinprodukte hoher Risikoklassen und in-vitro-Diagnostika sollen einer europäischen Bewertung unterzogen werden, wobei die Kriterien für die Auswahl der zu prüfenden Produkte und der Zeitrahmen vage formuliert sind.
Keine nationalen Verfahren mehr
Ist eine europäische Nutzenbewertung in Arbeit, darf kein Mitgliedsstaat dieselben wissenschaftlichen Auswertungen wiederholen. Allerdings können nach einer EU-Nutzenbewertung weiterhin zusätzliche Analysen gemacht werden, die für die nationale Entscheidungsfindung über die Erstattung notwendig sind.
Ein ganz wichtiger Punkt: Die eigentliche Erstattungsentscheidung und auch die Preisverhandlungen bleiben nationale Souveränität. Um in dieses Recht nicht einzugreifen, darf die Darstellung der wissenschaftlichen Ergebnisse keine Werturteile über den zusätzlichen klinischen Nutzen enthalten. Nur die nackten Fakten dürfen dargestellt werden.
Wie läuft das Verfahren ab?
Zur Durchführung des wissenschaftlichen Bewertungsprozesses für neue Therapien soll eine Koordinierungsgruppe etabliert werden, in der alle Mitgliedsstaaten vertreten sind. Sie soll den Bewertungsprozess steuern und durchführen. Die Kommission hat sich selbst im Verfahrensablauf allerdings weitgehende Eingriffsrechte gesichert.
Welche Evidenz zählt?
Die geringen Anforderungen an die von den Firmen einzureichenden Daten war einer der kritischsten Punkte in früheren Entwürfen. Sie orientierten sich am niedrigsten Standard der Mitgliedsstaaten und auf Modellbewertungen im Rahmen der freiwilligen europäischen Kooperation EUnetHTA.
EUnetHTA: Bewertungen von Herstellers Gnaden
EUnetHTA hat einige Bewertungen neuer Wirkstoffe durchgeführt, um ein gemeinsames europäisches Vorgehen zu erproben. Dass dabei die Firmen mit Samthandschuhen angefasst wurden, macht das Beispiel Sotagliflozin gegen Typ 1 Diabetes deutlich. Die Bewertung stellt Vorteile bei den Surrogatendpunkten „Senkung des Blutzuckers“ und „seltenere Unterzuckerungen“ fest. Gleichzeitig stellt sie in Frage, ob die leichten Verbesserungen klinisch überhaupt relevant sind. Das Risiko von Ketoazidosen wird als Nachteil benannt.
Vor allem aber wird die Aussagesicherheit der verwendeten Evidenz als „sehr niedrig“ bezeichnet.[7] Und das hat unmittelbar mit dem Verhalten des Anbieters Sanofi zu tun. Große Teile der eingereichten Evidenz konnten nicht verwendet werden, weil Sanofi einer Veröffentlichung nicht zustimmte: „Die Autoren durften die vertraulichen Informationen aus den Anlagen des eingereichten Dossiers nicht verwenden und haben diese Informationen auf Verlangen des Inhabers der Genehmigung für das Inverkehrbringen aus dem Dokument entfernt.“[8]
Es drohte eine Einigung auf den niedrigsten Standard: Nur die lückenhaften veröffentlichten Daten zu einem Arzneimittel wären berücksichtigt worden. Für eine seriöse Bewertung ist aber die Vorlage des Clinical Study Reports (CSR), der alle in einer klinischen Studie gemessenen Ergebnisse enthält, unentbehrlich. Das deutsche Gesetz zur Nutzenbewertung (AMNOG) schreibt genau das vor, viele andere EU-Staaten geben sich derzeit mit weniger zufrieden.
Der vorliegende EU-Entwurf verlangt ausdrücklich, dass das Dossier des Herstellers „alle veröffentlichte und unveröffentlichte Information, Daten, Analysen und andere Evidenz auf dem neuesten Stand“ enthalten müsse. Dazu gehören die (umfangreichen) Daten, die der Zulassungsbehörde EMA vorgelegt wurden. Auch das Schlüsselwort CSR wird ausdrücklich erwähnt, ebenso die Daten Dritter einschließlich abgebrochener Studien. All das ist ein entscheidender Fortschritt gegenüber allen bisher kursierenden Vorschlägen.
Als Unsicherheit bleibt, dass Einzelheiten über die vorzulegenden Unterlagen von der Kommission selbst in einem delegierten Rechtsakt festgelegt werden. Entscheidend wird die Gestaltung der Dossiervorlage (Template) sein, die den Herstellern genau vorschreibt, welche Daten in welcher Detailtiefe vorgelegt werden müssen.
So tun als ob?
In der bisherigen Debatte um die EU-Nutzenbewertung spielten auch sogenannte Surrogatendpunkte eine Rolle. Solche Laborwerte oder Messungen der Tumorgröße bei Krebs gelten als sehr unzuverlässiger Indikator für tatsächliche Vorteile.[9] Für PatientInnen wirklich relevant sind dagegen eine Senkung der Sterblichkeit, die Linderung der Symptome oder eine Verbesserung der Lebensqualität.
Im Gesetzentwurf ist jetzt von „gesundheitlichen Ergebnissen“ (health outcomes) die Rede. Das ist zwar eher allgemein gehalten, aber geht in die richtige Richtung. Die Praxis der kommenden EU-Bewertung wird zeigen, wie gut die relevanten Endpunkte und die PatientInnengruppen, die potenziell von einer Behandlung profitieren, definiert werden.
Zum Knackpunkt kann die Auswahl der Vergleichstherapie werden. Die Koordinierungsgruppe legt sie „unter Berücksichtigung von Informationen durch den Medikamentenentwickler, PatientInnen, klinische und andere ExpertInnen“ fest. Für den Hersteller ist der Vergleich mit einem neueren teuren Medikament möglicherweise die attraktivere Alternative, auch wenn für dieses Vorteile gegenüber älteren preisgünstigen Therapien gar nicht belegt sind. Denn so lassen sich am Ende eher hohe Erstattungspreise erzielen – selbst wenn das eigene Präparat keine Vorteile aufweisen sollte. Hier kommt es darauf an, dass die Koordinierungsgruppe stringent arbeitet und Beeinflussungsversuchen widersteht.
Die Mitgliedsstaaten können unterschiedliche Anforderungen zur Vergleichstherapie oder Endpunkten formulieren. Stimmt dem die Koordinierungsgruppe zu, werden mehrere Auswertungen parallel durchgeführt.
Real World Data?
Zwar ist im Entwurf die Rede von vergleichenden klinischen Studien als Goldstandard für die Bewertung. Es bleibt aber eine Hintertür offen, auch wissenschaftlich wenig belastbare Ergebnisse mit einzubeziehen. So ist im Begründungstext mehrfach von Real World Data die Rede, also nachträglichen Auswertungen von Daten aus der Behandlungspraxis. Damit sollen perspektivisch „Evidenzlücken gefüllt“ werden. Dafür sind solche Daten aber schon aus methodischen Gründen wenig geeignet. Erfreulicherweise taucht der Begriff Real World Data im Gesetzestext selbst aber nicht auf.
Eng getaktet
Bislang begann die Bewertung eines neuen Arzneimittels mit dem Markteintritt. Künftig soll sie früher starten, also deutlich vor der Zulassung. Das bedeutet mehr Unsicherheit, weil die klinischen Studien weniger weit gediehen sind. Für den Anbieter verspricht das europäische Verfahren schnelleren Marktzutritt und damit schnelleren Gewinn. Ein Versprechen wird das Verfahren allerdings nicht einlösen: Dass die neuen Medikamente überall verfügbar sein werden. Denn oft scheitert die Erstattung in ärmeren Mitgliedsstaaten auch an den hohen Preisen der neuen Mittel. Nicht selten vermarkten die Hersteller ihre Produkte in diesen Ländern erst gar nicht. Eine Verpflichtung, ein EU-weit zugelassenes Mittel auch überall anzubieten, enthält der Gesetzentwurf nicht.
Bereits deutlich vor der Zulassung muss die Koordinierungsgruppe die Vergleichstherapie und auch die zu bewertenden Aspekte bestimmen. Also zu einem Zeitpunkt, zu dem die genaue Indikation und eventuelle Anwendungsbeschränkungen noch nicht endgültig feststehen. Die Kommission fordert daraufhin die Unterlagen des Herstellers an. Wieviel Zeit ihm zur Erstellung des Dossiers zur Verfügung steht, ist noch nicht festgelegt. Klar ist lediglich, dass es bereits 112 Tage vor der geplanten Marktzulassung fertig sein muss.[10]
Genaue Ablaufpläne mit Fristen müssen Koordinierungsgruppe und Kommission erst noch entwickeln. Das wird erst nach Verabschiedung der EU-Verordnung geschehen. Alles in allem scheint der Zeitrahmen sehr eng, da das Abschlussdatum für den Bewertungsbericht mit 30 Tagen nach Zulassung fest terminiert ist.[11] Denn der für die Auswertung der Studienergebnisse zur Verfügung stehende Zeitraum wird durch mehrere Prozessschritte erheblich verkürzt. Die Kommission reicht das Herstellerdossier erst nach Prüfung auf Vollständigkeit „zügig“ an die Koordinierungsgruppe weiter. Dann müssen die BewerterInnen (Assesors)[12] zunächst einen vorläufigen Bericht erstellen, den PatientInnen und ExpertInnen kommentieren können.
Zensur durch Firmen?
Auch der Hersteller bekommt das Recht, den Entwurf zu sehen, und kann auf „alle rein technischen oder sachlichen Ungenauigkeiten“ hinweisen. Gleichzeitig kann er mitteilen, welche Informationen er als Geschäftsgeheimnisse betrachtet und geschwärzt sehen möchte. Das ist ein weitgehendes Eingriffsrecht, das Hersteller zum Beispiel im deutschen Nutzenbewertungsverfahren nicht haben. Hierzulande können sie erst die fertige bereits veröffentlichte Bewertung kommentieren und Schwärzungen gibt es gar nicht.
Die BewerterInnen müssen die Kommentare von PatientInnen, ExpertInnen und des Herstellers „abwägen“ und gegebenenfalls berücksichtigen. Erst dann können sie ihren Bericht abschließen, den die Koordinierungsgruppe noch gutheißen muss. Kommt keine Einigung zustande, werden die divergierenden Meinungen dargestellt.
Genehmigung durch Kommission
Abschließend prüft die EU-Kommission innerhalb von zehn Arbeitstagen, ob der Bericht den gesetzten Regeln entspricht. Ist das nicht der Fall, muss die Koordinierungsgruppe nachbessern. Findet der Bericht dann immer noch nicht das Wohlwollen der Kommission, teilt sie die Berichte und die Gründe für ihre Beanstandung nur den Mitgliedsstaaten und dem Hersteller mit. Die Öffentlichkeit erfährt von einem solchen Dissens erst nachträglich in einem zusammenfassenden Jahresbericht der Koordinierungsgruppe.
Wenn der Hersteller nicht liefert
Die Kommission entscheidet darüber, ob die vom Hersteller eingereichten Unterlagen für eine Nutzenbewertung den festgelegten Kriterien entsprechen und rechtzeitig eingereicht wurden. Kommt der Hersteller einer weiteren Aufforderung nicht nach, wird das Bewertungsverfahren eingestellt. Dann können nationale Bewertungen wieder uneingeschränkt durchgeführt werden.
Transparenz
Zwar heißt es im Begründungstext „Transparenz und Sensibilisierung der Öffentlichkeit für den Prozess sind von wesentlicher Bedeutung. Für den Fall, dass aus kommerziellen Gründen vertrauliche Daten vorliegen, muss die Vertraulichkeit klar definiert und begründet […] werden.“ Aber während des Bewertungsprozesses bleiben die Informationen vertraulich. Dritte, die die Daten kommentieren, müssen eine Geheimhaltungsvereinbarung unterschreiben.
Das Herstellerdossier und der Bewertungsbericht sollen „zum Zeitpunkt der Veröffentlichung“ auch auf der öffentlich zugänglichen Internetseite der EU-HTA zugänglich gemacht werden. Welche Teile des Herstellerdossiers veröffentlicht werden müssen, erschließt sich nicht so ohne weiteres. Im Entwurf ist aber die Rede von der „Entfernung von Informationen kommerziell sensibler Art“ vor Veröffentlichung. Dass der Hersteller auch im Bewertungsbericht selbst Schwärzungsvorschläge machen kann, wurde oben bereits erwähnt.
Was die frühe Beratung der Hersteller zu relevanten Endpunkten für seine Studien angeht, soll es auf der öffentlichen Website und im Jahresbericht der Koordinierungsgruppe eine „zusammenfassende anonymisierte Darstellung der nicht-vertraulichen Informationen“ geben. Im Jahresbericht der Koordinierungsgruppe einschließlich der eingeholten Kommentare von Externen.
Interessenkonflikte
Erfrischend deutlich heißt es im Entwurf: „Vertreter, die für die Koordinierungsgruppe oder ihre Untergruppen benannt werden und PatientInnen, klinische und sonstige Sachverständige, die an gemeinsamen Arbeiten teilnehmen, dürfen keine finanziellen oder sonstigen Interessen in der Industrie, die Gesundheitstechnologien entwickelt, haben, die ihre Unabhängigkeit oder Unparteilichkeit beeinträchtigen könnten.“ Die Kommission kann Mitglieder der Koordinierungsgruppe von einzelnen Diskussionen und dem Zugang zu den Unterlagen ausschließen, wenn sie deren Unabhängigkeit oder Unparteilichkeit nicht gewährleistet sieht. Die Erklärungen und eventuelle Ausschlüsse müssen in den Sitzungsprotokollen dokumentiert werden. Auch hier gilt die Einschränkung: Die genauen Kriterien, was einen Interessenkonflikt darstellt und wie damit umgegangen werden soll, werden von der Kommission erst später festgelegt.
Frühe Beratung
Eine wichtige Komponente der Nutzenbewertung ist die frühe Beratung der Hersteller, damit sie überhaupt geeignete Studien durchführen. Denn noch immer werden neue Medikamente mitunter nur gegen Placebo oder gar nichts[13] getestet.
Allerdings ist die individuelle Beratung einzelner Hersteller sehr kritisch zu sehen, da sie der Geheimhaltung unterliegt. Die BUKO Pharma-Kampagne fordert daher schon lange indikationsbezogene Beratungen, die öffentlich geführt werden können.[14] Das wäre nicht nur transparent, sondern würde auch wiederkehrende Beratungen zum selben Thema vermeiden. Immerhin ist im aktuellen Entwurf der Kritik der EU-Ombudsfrau an dem Prozedere für solche Beratungen bei der Zulassungsbehörde EMA[15] Rechnung getragen worden: Die Personen, die im Rahmen der EU-HTA beratend tätig waren, sollen an der späteren Bewertung desselben Produktes nicht beteiligt sein. Allerdings soll es begründete Ausnahmen geben, falls „die notwendige spezifische Expertise sonst nicht vorhanden“ sei.
Wer zahlt?
Erfreulicherweise ist eine Finanzierung der europäischen Nutzenbewertung durch Gebühren der Hersteller erst einmal vom Tisch. Die gesamten Kosten des Verfahrens werden aus Steuermitteln bezahlt. Allerdings soll nach drei Jahren evaluiert werden, ob nicht doch Gebühren erhoben werden sollen, um „die Unabhängigkeit der Koordinierungsgruppe sicherzustellen“. Als könne nicht gerade das die Unabhängigkeit gefährden: Kontrollen durch diejenigen finanzieren zu lassen, deren Produkte kontrolliert werden.
Der Gesetzentwurf soll dem EU-Parlament noch vor Ende des Jahres zur endgültigen Entscheidung vorgelegt werden. (JS)
Artikel aus dem Pharma-Brief 7/2021, S.1
[1] Pharma-Brief (2018) Wunschkonzert für Hersteller. Nr. 3, S. 1
[2] Pharma-Brief (2018) Zwischen Kommerz und Transparenz. Nr. 6, S. 5
[3] Pharma-Brief (2018) EU-HTA Update 2. Nr. 7, S. 6
[4] Draft text for a regulation of the European Parliament and of the Council on health technology assessment and amending Directive 2011/24/EU www.europarl.europa.eu/meetdocs/2014_2019/plmrep/COMMITTEES/ENVI/DV/2021/07-12/Consolidated_clean_text_HTA_210630_post_Coreper_Rev_EN.pdf [Zugriff 27.8.2021]
[5] In anderen EU-Staaten werden viele Medikamente wegen fehlender oder zu geringer Vorteile bei hohen Preisen gar nicht erst in die Erstattung aufgenommen. In Deutschland können alle neue Medikamente bis auf sehr wenige Ausnahmen (Lifestyle-Arzneimittel) vom ersten Tag der Vermarktung an zu Lasten der Krankenkassen verschrieben werden. Ein anschließender Ausschluss aus der Erstattung wegen Unwirtschaftlichkeit ist eher die Ausnahme, aber starke Preissenkungen bei fehlendem Zusatznutzen sind häufig.
[6] Formell heißt ein EU-Rechtsakt, der unmittelbar unionsweit gilt, Verordnung (Regulation) – im Gegensatz dazu müssen EU-Richtlinien noch in nationale Gesetze umgesetzt werden.
[7] EUnetHTA (2019) Relative effectiveness assessment of pharmaceutical technologies. Sotagliflozin is as an adjunct to insulin therapy to improve glycaemic control in adults with type 1 diabetes mellitus with a body mass index (BMI) ≥ 27 kg/m2, who have failed to achieve adequate glycaemic control despite optimal insulin therapy. www.eunethta.eu/wp-content/uploads/2019/06/PTJA04-Final-Assessment-Report-V1.pdf [Zugriff 8.9.2021]
[8] EUnetHTA (2019) Input from manufacturer on the 2nd draft assessment “Sotagliflozin for adult patients with type 1 diabetes mellitus and with a body mass index (BMI) ≥ 27 kg/m2 who have inadequate glucose control using optimised insulin or insulin analogues” https://eunethta.eu/wp-content/uploads/2019/06/PTJA04-Comments-MAH-factcheck- V1.pdf [Zugriff 8.9.2021]
[9] Pharma-Brief (2017) Bescheidener Fortschritt. Nr. 7-8, S. 1
[10] 45 Tage vor der geplanten Stellungnahme des Ausschusses für Humanarzneimittel (CHMP[10]) der europäischen Zulassungsbehörde EMA. Diese Stellungnahme des CHMP ist die eigentliche inhaltliche Weichenstellung für ein neues Arzneimittel. Formal entscheidet die Kommission dann spätestens 67 Tage danach über die Marktzulassung.
[11] Die Koordinierungsgruppe muss die Bewertung des Herstellerdossiers 30 Tage nach Erteilung der Zulassung abgeschlossen haben. Stehen also für den gesamten Prozess theoretisch 142 Tage (112+30) zur Verfügung.
[12] Die Koordinierungsgruppe wählt einen Bewerter und einen Co-Bewerter aus, die aus unterschiedlichen Mitgliedsstaaten stammen müssen.
[13] Einarmige Studien ohne jeden direkten Vergleich
[14] Pharma-Brief (2017) In Watte gepackt. Nr. 10, S. 1
[15] Pharma-Brief (2018) Europa: Undurchsichtige Beratung. Nr. 4-5, S. 16
EU ordnet Preissenkungen für Blutkrebs-Präparate an
EU-Maßnahme gegen Aspen lenkt den Blick auf globale Preishürden
Die EU-Kommission hat Anfang Februar die Verpflichtungszusagen der Firma Aspen Pharmacare für bindend erklärt. Der jahrelange Streit mit dem Hersteller um extreme Kosten und geringe Verfügbarkeit von sechs Arzneimitteln soll damit beigelegt werden.
Konkret geht es um die Wirkstoffe Melphalan, Chlorambucil, Mercaptopurin, Tioguanin und Busulfan, die bei der Behandlung verschiedener Formen von Blutkrebs eingesetzt werden. Aspen hatte die Herstellung der Präparate 2012 nach Auslauf des Patentschutzes übernommen – und anschließend die Preise nach oben gejagt. Die EU antwortete 2017 mit einem Prüfverfahren und warf dem Hersteller vor, seine Marktmacht zu missbrauchen: „Selbst nach Berücksichtigung einer angemessenen Rendite hätten die Preise durchschnittlich um fast 300 Prozent über den relevanten Kosten gelegen, so die Kommission, wobei der Überschuss von Produkt zu Produkt und von Land zu Land unterschiedlich groß ausgefallen sei.“[1] Deutschland war als eines der ersten europäischen Länder von den Preissprüngen betroffen.
Bewusste Verknappung
Die Firma setzte darauf, dass in vielen Fällen schlichtweg keine oder nur wenige Alternativen zur Verfügung standen. Als sie Gegenwind erhielt, griff sie laut EU zu noch härteren Mitteln: „Als nationale Behörden versuchten, sich den Preiserhöhungen zu widersetzen, drohte Aspen gar, die Medikamente aus den nationalen Listen erstattungsfähiger Arzneimittel streichen zu lassen, und gab in einigen Fällen sogar seine Absicht bekannt, die üblichen Lieferungen in den betreffenden Mitgliedstaaten einzustellen.“[2]
Die EU machte jetzt Ernst: Die Firma muss die Preise der sechs Medikamente im Schnitt um 73% senken, auf das Niveau von 2012. Die Preise gelten rückwirkend vom 1. Oktober 2019 für zehn Jahre, zum Ausgleich muss Aspen eine Einmalzahlung leisten. Der Hersteller musste zudem die Versorgung für die EU in den kommenden fünf Jahren zusichern. Bei Verstößen droht ein Bußgeld von bis zu 10% vom Gesamtumsatz der Firma. Nach eigenen Angaben machte Aspen zuletzt einen Jahresumsatz von fast zweieinhalb Milliarden US-Dollar.
Der Skandal wirft nicht nur ein grelles Schlaglicht auf weit verbreitete, skrupellose Praktiken der Pharma-Industrie im Milliardenmarkt mit Krebs-Präparaten. Er zeigt auch die globale Dimension des Problems.
Dramatische Lage in Südafrika
Aspen besitzt zwar mehrere Tochtergesellschaften in Europa, hat seinen Sitz jedoch in Südafrika. Auch dort zeigte der Clinch mit der EU-Kommission Folgen. Das machte Salomé Meyer, Aktivistin und Projektmanagerin bei der südafrikanischen Cancer Alliance im Gespräch mit der Pharma-Kampagne deutlich.[3] Mit Blick auf die beanstandeten Präparate stellt sie fest: „Einige der Produkte werden bei uns im öffentlichen Sektor schon gar nicht mehr zur Behandlung eingesetzt. Melphalan z.B., das zur Therapie von Myelom-PatientInnen benötigt wird, wurde über das staatliche Ausschreibungsverfahren für 2020-2022 nicht erworben. Angeblich aufgrund von Produktionsengpässen des Herstellers.“ Meyer vermutet jedoch einen anderen Grund: „Es kann durchaus sein, dass dies eher mit der EU-Untersuchung zusammenhing, als mit tatsächlichen Problemen in der Fabrikation.“
In Südafrika nimmt die Anzahl an Krebserkrankungen deutlich zu. 2018 starben daran laut Weltgesundheitsorganisation über 57.000 Menschen.[4] Die Versorgungslage mit den notwendigen Medikamenten ist schlecht. Hohe Preise und Patente spielen dabei eine elementare Rolle, so die Cancer Alliance. Die zivilgesellschaftliche Organisation hat in den letzten Jahren die Zugangshürden bei vielen Krebsmedikamenten analysiert.[5]
EU-Wettbewerbskommissarin Margrethe Vestager sagte zum Beschluss gegen Aspen, er sei „eine klare Botschaft an andere marktbeherrschende Pharmaunternehmen, keine missbräuchlichen Preisbildungspraktiken anzuwenden, mit denen unsere Gesundheitssysteme ausgenutzt werden.“[6] Es wäre allerdings wünschenswert, wenn die EU dabei auch Hersteller aus der eigenen Wirtschaftsregion und deren Vermarktungspraktiken in armen Ländern ins Auge fassen würde. (MK)
Projekt „Unbezahlbar krank?“: Mit ihrem Projekt macht die Pharma-Kampagne auf die schlechte Verfügbarkeit von Krebsmedikamenten aufmerksam – gerade in Ländern des globalen Südens. Im April wird ein Pharma-Brief Spezial zum Thema erscheinen. Er soll die Unterversorgung in Südafrika, Tansania, Äthiopien, Ecuador und Indien beleuchten. Dazu steht die Pharma-Kampagne in engem Austausch mit ExpertInnen vor Ort.
Artikel aus dem Pharma-Brief 2/2021, S.7
[1]DAZ (2021) Aspen senkt Preise für sechs Krebsarzneimittel. www.deutsche-apotheker-zeitung.de/news/artikel/2021/02/10/aspen-senkt-preise-fuer-sechs-krebsarzneimittel [Zugriff 12.02.2021]
[2] Chemiereport (2021) Aspen muss Preise massiv senken. www.chemiereport.at/aspen-muss-preise-massiv-senken. [Zugriff 12.02.2021]
[3]Gespräch mit Salomé Meyer am 11.02.2021
[4]WHO (2020) Cancer Country Profile 2020: South Africa. www.who.int/cancer/country-profiles/ZAF_2020.pdf?ua=1 [Zugriff 12.02.2021]
[5]Cancer Alliance (2018) Exploring patent barriers to cancer treatment access in South Africa: 24 medicine case studies. https://canceralliance.co.za/wp-content/uploads/2019/08/Exploring-Patent-Barriers-to-Cancer-Treatment-Access-in-SA-24-Medicine-Case-Studies-October-2017-update-January-2018.pdf [Zugriff 12.02.2021]
[6]Europäische Kommission (2021) Kartellrecht: Kommission akzeptiert Verpflichtungszusagen von Aspen gegen exzessiv hohe Preise für patentfreie Krebsmittel. https://ec.europa.eu/germany/news/20210210-kartellrecht-aspen_de [Zugriff 12.02.2021]
Die Macht der globalen Gesundheitsdaten
„Institute for Health Metrics and Evaluation“ in der Kritik
Für effektive Gesundheitsinterventionen sind verlässliche Daten über Krankheitshäufigkeit und -verbreitung enorm wichtig. Dass mit dem Institute for Health Metrics and Evaluation (IHME) eine private Einrichtung zum globalen Meinungsführer wurde, ist nicht unproblematisch.
Die internationale Sammlung von Daten zur gesundheitlichen Lage der Welt ist eigentlich bei der Weltgesundheitsorganisation (WHO) verortet. Ausgerechnet zwei ehemalige Mitarbeiter der WHO, Christopher J L Murray und Alan D Lopez, gründeten das IHME.[1] Sie begründeten ihren Schritt mit einer ausdrücklichen Kritik an der Zuverlässigkeit der von der WHO gesammelten Daten: Diese seien manipulationsanfällig, weil die Mitgliedsstaaten öfters versuchten, ihre gesundheitliche Lage rosiger darzustellen als sie in Wirklichkeit sei.[2] Auch wenn das nicht völlig von der Hand zu weisen ist, bleibt die Frage, ob die Privatisierung dieser öffentlichen Aufgabe die beste Lösung ist.
2007 wurde das IHME mit massiver Unterstützung der Bill und Melinda Gates Foundation aus der Taufe gehoben. Es ist an der University of Washington in Seattle, USA, angesiedelt. Zentrales Projekt: Die Global Burden of Disease Study, die versucht, die Krankheitslast auf der Welt möglichst genau zu erfassen und sich nicht auf die Zählung von Todesfällen zu beschränken. Das war allerdings nichts Neues. Ursprünglich war die systematische Erfassung der Global Burden of Disease (GBD) ein Konzept, das von der Weltbank in den 1990er-Jahren gemeinsam mit der WHO – unter Beteiligung von Murray – vorangetrieben wurde. Die WHO veröffentlicht Daten zur weltweiten Krankheitslast seit dem Berichtsjahr 2000. [3],[4]
Das IHME publizierte seine erste Global Burden of Disease Study 2012. Das war durchaus als Konkurrenz zur WHO zu verstehen. Die WHO hielt die Ergebnisse dieser Studie teils nicht für vertrauenswürdig, weil sie die Informationen zur Methodik für unzureichend hielt.[4] Inzwischen gelten die Publikationen des IHME als Standard für globale Gesundheitsdaten – und die WHO wurde an den Rand gedrängt. Seit 2018 kooperiert die WHO mit dem Institut.[1] Allein von der Gates-Stiftung hat das IHME über die Jahre über 600 Millionen US$ an Förderung erhalten.
Tim Schwab fasste die Kritik am IHME kürzlich in „The Nation“ prägnant zusammen.[5] Zwar sei es lobenswert, zu besseren Daten kommen zu wollen, aber das IHME nutze teils schwer nachvollziehbare Methoden, um fehlende Daten durch Simulationen zu ersetzen. Da die Ergebnisse des IHME aber zunehmend als Basis für Entscheidungen staatlicher und privater Träger über den Einsatz von Geldern für die Gesundheitsversorgung dienen, sei das nicht unproblematisch.
Win-win für wen?
Durch die Kooperation mit der angesehenen Medizinzeitschrift Lancet beim Global Burden of Disease Report prägt das IHME immer stärker den öffentlichen Diskurs. Dabei sind sowohl das Institut als auch die Verleger des Lancet Gewinner. Durch zahlreiche Artikel des IHME in der Fachzeitschrift, die häufig zitiert werden, steigert sich der „Impact Factor“ des Lancet.
2019 erhielt der Herausgeber des Lancet, Richard Horton, den mit 100.000 US$ dotierten Roux-Preis des IHME. Ein Mitarbeiter des Instituts klagte in einer internen E-Mail: „Ich würde gerne verstehen, was der langfristige Denkprozess für die Verleihung des Preises an Horton war. Wie sollen wir als MitarbeiterInnen diese Entscheidung verteidigen, wenn uns vorgeworfen wird, dass wir uns beim Lancet eingekauft haben, statt dass unsere Artikel wegen der Qualität unserer Arbeit veröffentlicht werden?“[5]
Nicht wenige WissenschaftlerInnen beklagen eine Hegemonie des IHME bei der Erfassung und Interpretation von internationalen Gesundheitsdaten.
Alle seriösen Fachzeitschriften in der Medizin verlangen eine Offenlegung von Interessenkonflikten. Das IHME scheint es damit nicht so genau zu nehmen. Dass das Institut 2018 über drei Millionen US$ von der Pharmaindustrie kassiert hat, taucht unter den Artikeln des Instituts nicht auf.[5]
Dass das IHME tatsächlich nicht immer richtig liegt, zeigte sich zu Beginn der Covid-19-Pandemie im Frühjahr 2020. Die weit in die Zukunft reichenden Prognosen für den Verlauf enthielten eine deutliche Unterschätzung der Erkrankungszahlen. Sie dienten dem damaligen US-Präsidenten Trump als Rechtfertigung, keine einschneidenden Maßnahmen zu veranlassen.[5] (JS)
Artikel aus Pharma-Brief 1/2021, S. 4
[1] Shiffman J and Shawar YR (2020) Strengthening accountability of the global health metrics enterprise. Lancet; 395, p 1452 https://doi.org/10.1016/S0140-6736(20)30416-5
[2] Murray CJL et al. (2004) Monitoring global health: time for new solutions Christopher J L, Alan D, Suwit BMJ; 329, p 1096
[3] WHO (2021) Global Health Estimates (GHE) www.who.int/healthinfo/global_burden_disease/en/
[4] WHO (2017) WHO methods and data sources for global burden of disease estimates 2000-2015. www.who.int/healthinfo/global_burden_disease/GlobalDALYmethods_2000_2015.pdf
[5] Schwab T (2020) Are Bill Gates’s Billions Distorting Public Health Data? The Nation, 6 Dec. www.thenation.com/article/society/gates-covid-data-ihme [Zugriff 24.1.2021]
Covid-Patentpool: Neustart oder Strohfeuer?
Einjähriges Bestehen vom C-TAP der WHO
Ursprünglich sollte der Patentpool C-TAP[1] freiwillige Lizenzen und Know-how von Herstellern einholen, um die Produktion von Impfstoffen und Medikamenten global auszuweiten und so die Covid-19-Bekämpfung voranzubringen. Doch das Projekt der Weltgesundheitsorganisation (WHO) konnte in den letzten zwölf Monaten keine Erfolge vermelden. Nun versucht die Organisation einen Neustart. Denn die Produktion von mehr Impfdosen für den globalen Süden kommt wegen politischer Blockaden nicht voran.
Kurz vor den Sommerferien dreht sich ein gewichtiger Teil der bundesdeutschen Covid-19-Debatte um Urlaubsfragen – mit welchen Einschränkungen in verschiedenen Bundesländern oder dem Ausland zu rechnen ist oder wie es um einen digitalen Impfpass bestellt sein wird. Im krassen Kontrast dazu stehen die Nachrichten aus dem Rest der Welt. So trat etwa Argentinien wegen einer zweiten Welle von Covid-19 bis Monatsende in einen harten Lockdown.[2] Währenddessen meldeten vietnamesische Stellen, man habe eine Mutation entdeckt, die eine Kombination aus Alpha- und Delta-Variante darstelle (ehemals „britische“ bzw. „indische“ Variante).[3] In Indien wiederum eskalierte die epidemiologische Lage derart, dass das Land im April einen vorübergehenden Exportstopp von Covid-19-Impfstoffen verkündete – mit massiven Folgen auch für die Belieferung des afrikanischen Kontinents.[4]
COVAX schwächelt, Waiver stockt
Bis Anfang Mai wurden weltweit insgesamt 1,1 Milliarden Covid-19-Dosen eingesetzt, gerade einmal 0,3% davon wurden in Ländern mit niedrigem Einkommen verimpft.[5] Es passt ins Bild, dass die Verteilungsplattform COVAX, auf die auch die deutsche Regierung primär setzt, weiterhin kaum Wirkung zeigt. Zeitgleich nimmt der Druck auf die Welthandelsorganisation (WTO) zu, den seit Oktober 2020 schwelenden Streit um einen „Patent-Waiver“ (wir berichteten[6]) endlich konstruktiv anzugehen.
Mittlerweile hat die US-Regierung zumindest bezüglich Impfstoffpatenten eine Unterstützung für die Initiative Indiens und Südafrikas zur vorübergehenden Aussetzung der geistigen Eigentumsrechte für Covid-19-Produkte signalisiert. Dieser Waiver wird von weit über 100 WTO-Mitgliedern direkt oder indirekt getragen. Da zu dieser Gruppe mittlerweile auch China und Russland zählen,[7] steht nun die EU im Fokus: Während die Kommission weiterhin mauert, votierte das europäische Parlament Ende Mai für Ausnahmeregelungen für Covid-19-Impfstoffe.[8] Die Bundesregierung gilt weiterhin als einer der wichtigsten Bremser innerhalb der EU.[9]
Wissenschaft für Waiver
Auch die Stimmen aus der Wissenschaft für den Waiver mehren sich. In Großbritannien konstatierte ein Arbeitspapier der renommierten London School of Economics: „Der TRIPS-Waiver ist in diesem Zusammenhang ein wesentliches Rechtsinstrument, um eine radikale Erhöhung der Produktionskapazitäten und damit des Angebots an COVID-19-Impfstoffen zu ermöglichen und so einen Weg zu einem weltweiten gerechten Zugang zu schaffen.“ [10] Hierzulande argumentierte unter anderem der Präsident des Deutschen Instituts für Wirtschaftsforschung (DIW) für die WTO-Initiative und wandte sich dabei auch gegen die Mär der Innovationsfeindlichkeit: „Im Gegenteil, der Wettbewerb und damit auch die Innovation würde zunehmen. Zumal die mRNA-Technologie nicht nur als Impfstoff gegen Covid, sondern für viele andere Krankheiten, vor allem Krebs, erforscht wird und erste Erfolge gezeigt hat.“ [11]
Das jüngste TRIPS Council-Meeting der WTO (8./9. Juni) fand erst nach Drucklegung statt. Eines ist allerdings sicher: Für eine global gerechtere Versorgung mit Covid-19-Impfstoffen ist die Ausweitung von Produktionskapazitäten elementar. Dafür ist Technologietransfer und das Teilen von Know-how vonnöten. Denn auch wenn die Herstellung in den kommenden Monaten wohl anziehen wird, bleibt die Versorgungslage global gesehen prekär und der Markt extrem umkämpft.[12]
C-TAP: Trauriger Geburtstag
Um ein wichtiges Instrument für mehr Impfstoff-Produktion war es erstaunlicherweise lange verdächtig still: Den WHO Patentpool für Covid-19, C-TAP. Bereits im Mai 2020 lanciert, beging das Konstrukt zuletzt seinen einjährigen Geburtstag. Grund zum Feiern gab es nicht, denn der Status quo ist niederschmetternd: Auch nach zwölf Monaten haben Hersteller keine einzige Lizenz eingebracht. Die Situation des C-TAP kann damit als sinnbildlich für die dramatische Verschleppung in der globalen Covid-19-Bekämpfung gelten. Obwohl ein Mittelweg zwischen dem strukturellen Ansatz des WTO-Waivers und der eher symbolischen Mildtätigkeit von COVAX,[13] wurde er von Pharmaindustrie und international einflussreichen Staaten (darunter Deutschland) weitestgehend ignoriert. Statt nicht-exklusiven und transparenten Lizenzvereinbarungen, wie C-TAP sie anstrebt,[14] forcieren die reichen Länder angesichts der nicht mehr zu übersehenden globalen Ungerechtigkeiten erst jetzt freiwillige Vereinbarungen zwischen Herstellern und ärmeren Ländern. Doch das ist eine stark limitierte Variante des Zugangs, die zu weiteren Verzögerungen und zu ungerechtfertigt hohen Preisen führt.
Die Schwäche von COVAX und die wachsende Unterstützung für die WTO-Ausnahmeregelung könnten nun allerdings den Blick auf C-TAP neu schärfen. Jedenfalls versucht sich die WHO darin, einen frischen Impuls für die Initiative zu setzen. Eine Pressekonferenz am 28. Mai sollte dabei ein erster Schritt sein.
Gute News nur im Kleinen
In dieser Veranstaltung sprach neben WHO Generaldirektor Thedros Ghebreyesus auch der Präsident Costa-Ricas, Carlos Alvarado Quesada. Das zentralamerikanische Land hatte im vergangenen Jahr den Start von C-TAP maßgeblich mit initiiert. Wer allerdings bei der Wiederbelebung des Projekts auf große Schlagzeilen gehofft hatte, wurde durch die Veranstaltung enttäuscht. Positive Neuigkeiten gab es nur im Kleinen:
Mit Spanien konnte C-TAP immerhin zusätzlich zu den offiziell unterstützenden 42 Staaten ein weiteres Mitglied gewinnen. Auch wurde von WHO-Seite kommuniziert, dass man momentan mit verschiedenen Produzenten in Gesprächen sei, die möglicherweise Lizenzen einbringen würden: Zwei davon beträfen Covid-19-Impfstoffe und fünf entsprechende Diagnostika. Um welche Hersteller es sich handelt und was der konkrete Stand der Verhandlungen ist, wurde allerdings nicht verraten. Eher gering war daher auch das mediale Interesse an dem Geburtstags-Event: Erst die allerletzte Nachfrage in der abschließenden Presserunde bezog sich überhaupt direkt auf die Arbeit von C-TAP.
Tödliche Abhängigkeit
In der monatelangen Debatte um gerechten Zugang zu medizinischen Produkten gegen Covid-19 wurden viele argumentative Haken geschlagen. Der entscheidende Punkt hat sich besonders beim Thema Impfstoffe aber herauskristallisiert: Es geht letztlich um die tödliche globale Abhängigkeit vom Gutdünken einiger sehr großer Firmen aus dem globalen Norden und dem ausgeprägten Unwillen vieler Regierungen aus Ländern mit starker Pharmaindustrie, die daraus resultierenden frappierenden Ungerechtigkeiten ernsthaft anzugehen.
Und so werden im politischen Diskurs des globalen Nordens nicht progressive Lösungen und damit verbundene Chancen betont, sondern angebliche Risiken. Ein Beispiel ist die von DIW-Seite erwähnte mögliche Verbreitung von mRNA-Technologien. Statt positiv hervorzuheben, dass diese Proliferation nachhaltig regionale Forschungs- und Produktionsmöglichkeiten bei zukünftigen Pandemien – aber auch gegen nicht-übertragbare Erkrankungen – stärken könnte, werden vor allem mögliche Verluste für heimische Pharmafirmen moniert.
„Haltet den Dieb“
Wie dreist in diesem Diskurs mithin Ursache und Wirkung verdreht werden, zeigte zuletzt formvollendet Thomas Cueni, Generaldirektor des internationalen Pharmaverbands Ifpma, der Industrieländer ermahnte: „Wenn wirklich jetzt etwas getan werden soll, müssen reiche Staaten dazu bewegt werden, ihre Dosen zu teilen.“ [15] Als hätte die Industrie nicht durch ihre gierige Preispolitik und bewusste Verknappung der Produktionsmöglichkeiten von Impfstoffen selbst ganz maßgeblich an der globalen Schräglage mitgewirkt und dabei unglaublich abgesahnt (siehe unseren Beitrag über Covid-19 Milliardäre).
Wenigstens scheint das von Beginn an fadenscheinige Argument, der globale Süden hätte gar keine Kapazitäten für Covid-19-Impfstoffproduktion, endlich zermürbt worden zu sein. So erklärten beispielsweise während der C-TAP-„Feierlichkeiten“ Bangladesch und Indonesien, über erhebliche Produktionskapazitäten zu verfügen – wobei selbstverständlich die Freigabe von geistigen Eigentumsrechten, Technologie- und Know-how-Transfer wichtig sei.[16]
Auch das Argument, mRNA-Impfstoffe seien besonders schwer herzustellen, ist falsch. Suhaib Siddiqi, ehe-maliger Direktor für Chemie bei Moder-
na, sagte laut AP: „Mit einer Blaupause und technischem Rat sollte eine moderne Fabrik in der Lage sein, die Impf-
stoffproduktion in höchstens drei bis vier Monaten in Gang zu bringen.“ [17]
Verschenkte Zeit, verlorene Leben
Die Vehemenz, mit der von Teilen der Medien und Politik auch hierzulande gegen etwaige Lockerungen im Bereich geistigen Eigentums geschossen wird, verweist deutlich auf die Lobby-Kraft der Pharmaindustrie und die Tatsache, dass viele Akteure die Unantastbarkeit von Patenten ideologisch verinnerlicht haben. Dass es sich bei Patentrechten lediglich um einen (nur zum geringen Teil verwirklichten) gesellschaftlichen Vertrag zur Förderung medizinisch wichtiger Innovation handelt, scheint in Vergessenheit geraten zu sein.
Dem zum Trotz würde man sich wünschen, dass zumindest freiwillige Mechanismen in dieser weltweiten Ausnahmesituation greifen könnten. Der aktuelle Zustand des Covid-19-Patentpools zeigt jedoch abermals, dass „freiwillige Selbstverpflichtungen“ im globalen Gesundheitskapitalismus bloße Augenwischerei sind. Denn seit Gründung von C-TAP vor einem Jahr ist nicht nur kostbare Zeit verschenkt worden. Es sind auch viele Tausend Leben weltweit verloren gegangen. (MK)
Artikel aus dem Pharma-Brief 5/2021, S.1
[1] C-TAP: COVID-19 Technology Access Pool der WHO
[2] DW (2021) Argentina prepares for strict lockdown as Covid cases soar. www.dw.com/en/argentina-prepares-for-strict-lockdown-as-covid-cases-soar/a-57610622 [Zugriff 21.5.2021]
[3] Reuters (2021) Vietnam detects hybrid of Indian and UK COVID-19 variants. www.reuters.com/world/asia-pacific/vietnam-detects-hybrid-indian-uk-covid-19-variant-2021-05-29/ [Zugriff 31.5.2021]
[4] DW (2021) Corona: Afrikas Impfprogramm kommt nicht voran. 19. Mai www.dw.com/de/corona-afrikas-impf-programm-kommt-nicht-voran/a-57574684 [Zugriff 31.5.2021]
[5] WHO (2021) Director-General‘s opening remarks at the media briefing on COVID-19 – 14 May 2021 www.who.int/director-general/speeches/detail/director-general-s-opening-remarks-at-the-media-briefing-on-covid-19-14-may-2021
[6] Pharma-Kampagne (2021) Der „Dritte Weg“ ist eine Sackgasse. www.bukopharma.de/de/newsarchiv/450-kritik-an-wto-fuehrung [Zugriff 31.5.2021]
[7] Laskar RH (2021) Brics backs Covid-19 vaccine patent waivers. Hindustan Times 2 June www.hindustantimes.com/india-news/brics-backs-vaccine-patent-waivers-101622574659675.html
[8] Ärzteblatt (2021) EU-Parlament unterstützt Aussetzung von Patenten für Coronaimpfstoffe. 20. Mai www.aerzteblatt.de/nachrichten/124007/EU-Parlament-unterstuetzt-Aussetzung-von-Patenten-fuer-Coronaimpfstoffe [Zugriff 31.5.2021]
[9] Usher DJ (2021) South Africa and India push for COVID-19 patents ban. Lancet; 396, p 1790 www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)32581-2/fulltext [Zugriff 28.5.2021]
[10] Thambisetty S et al. (2021) The TRIPS Intellectual Property Waiver Proposal: Creating the Right Incentives in Patent Law and Politics to end the COVID-19 Pandemic. LSE Legal Studies Working Paper https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3851737 [Zugriff 31.5.2021]
[11] Fratzscher M (2021) Was für eine Freigabe der Impfstoff-Patente spricht. 10. Mai www.welt.de/debatte/kommentare/article231026345/Marcel-Fratzscher-Was-fuer-Freigabe-der-Impfstoff-Patente-spricht.html
[12] Kupferschmidt K and Cohen J (2021) Rich countries cornered COVID-19 vaccine doses. Four strategies to right a “scandalous inequity”. 26 May www.sciencemag.org/news/2021/05/rich-countries-cornered-covid-19-vaccine-doses-four-strategies-right-scandalous [Zugriff 28.5.2021]
[13] COVAX plant Impfdosen für nur 20% der Bevölkerung in ärmeren Ländern zur Verfügung zu stellen, die Auslieferung soll sich bis weit ins nächste Jahr hinziehen.
[14] WHO (2021) C-TAP. Enhancing global manufacturing capacity to address today’s and tomorrow’s pandemics. 16 Jan www.who.int/publications/m/item/c-tap-enhancing-global-manufacturing-capacity-to-address-today-s-and-tomorrow-s-pandemics [Zugriff 31.5.2021]
[15] Byatnal A (2021) The battle for COVID-19 vaccine IP rights: “We need all hands on deck”. Devex 26 May www.devex.com/news/the-battle-for-covid-19-vaccine-ip-rights-we-need-all-hands-on-deck-99985 [Zugriff 28.5.2021]
[16] Cullinan K (2021) Indonesia and Bangladesh reveal massive untapped vaccine production capacity at C-TAP anniversary. Health Policy Watch 28 May https://healthpolicy-watch.news/indonesia-and-bangladesh-reveal-massive-untapped-vaccine-production-capacity-at-c-tap-anniversary/ [Zugriff 31.5.2021]
[17] Cheng M and Hinnant L (2021) Countries urge drug companies to share vaccine know-how. AP News 1 March https://apnews.com/article/drug-companies-called-share-vaccine-info-22d92afbc3ea9ed519be007f8887bcf6 [Zugriff 3.6.2021]
Covid-19: Solidarität dringend gesucht
Globale Impfstoffversorgung in der Sackgasse?
Mit dem Covid-19-Impfstart in einigen Ländern wird der Blick auf die Verteilungspraxis kritischer. Dabei rücken besonders die verschiedenen Initiativen bei der WHO und der WTO in den Fokus.
Während in Deutschland eine Debatte darüber tobt, wann in den kommenden Wochen die notwendigen Impfstoffmengen gegen Covid-19 zur Verfügung stehen, stellt sich für den Großteil des globalen Südens die Frage, wie viele Monate oder Jahre vergehen mögen, bis es ausreichenden Zugang gibt.
Covid-19: Globales Versagen
Warum werden Profite statt Menschen geschützt?
Vor zwei Jahren wurde Covid-19 entdeckt, seit einem Jahr gibt es Impfstoffe. Doch große Teile der Weltbevölkerung sind vom Zugang ausgeschlossen. Konstruktive Vorschläge, die Produktion zügig auszuweiten, werden von wenigen Industrieländern blockiert, Deutschland gehört dazu. Jetzt rächt sich die global geringe Impfrate. Immer neue Virusvarianten machen auch vor reichen Ländern nicht halt.
Bereits die Delta-Variante des Virus war deutlich ansteckender als vorherige Covid-19 Typen. Sie verbreitete sich innerhalb kurzer Zeit weltweit. Seit kurzem ist die zuerst in Südafrika identifizierte Omikron-Variante in Europa angekommen. Noch ist unklar, welche Auswirkungen sie hat. Fest steht allerdings, dass eine schnelle Eindämmung der Pandemie wesentlich von hohen Impfraten überall auf der Welt abhängt. Dafür braucht es viele Impfdosen. Und die fehlen, denn die Patentinhaber beharren auf ihrem Recht, allein zu entscheiden, wo und wieviel Impfstoff produziert wird – obwohl die Entwicklung auf öffentlichen Erfindungen basiert und mit Milliardensummen für Forschung und Abnahmeversprechen abgesichert wurde.
Geradezu zynisch wirkt das Argument von Firmen und Politik, irgendwann im nächsten Jahr würde ja genügend Impfstoff produziert. Weder ist es sicher, dass das Versprechen Wirklichkeit wird, noch wird der Preis angemessen sein. Und letzteres ist auch von großer Bedeutung, da die Beschaffung von Impfstoffen über den globalen Verteilmechanismus COVAX mit öffentlichen Geldern finanziert werden muss.
„In der langen Geschichte der Pharmakonzerne ist ihr wichtigstes Anliegen, die Gewinne zu maximieren. Das ist ihr Geschäftsmodell. Um Gewinne zu maximieren, muss man das Angebot verknappen.“[1] - Wirtschaftsnobelpreisträger Josef Stieglitz
Das Herstellungsmonopol dient lediglich dazu, die Produktion zu beschränken und so möglichst hohe Preise zu sichern. Pfizer, Biontech und Moderna erzielen mit den Impfstoffen dieses Jahr nach Berechnungen der People's Vaccine Alliance einen Gewinn (!) von 34 Milliarden US$.[2]
Vorschläge lagen auf dem Tisch
Bereits im Mai 2020 brachte Costa Rica in die Weltgesundheitsversammlung einen sinnvollen Vorschlag ein: Man solle den für HIV-Medikamente existierenden Patentpool (MPP) auch für die damals noch zu entwickelnden Covid-19 Produkte zu nutzen und um Technologietransfern zu erweitern (C-TAP). Der Pool blieb aber bis vor einem Monat völlig leer,[3] kein einziger Impfstoff ist an den MPP lizensiert. Denn selbst diese (freiwillige) Lizensierung an den Patentpool wurde von reichen Ländern nicht unterstützt. Im Oktober 2020 stellten daher Indien und Südafrika den Antrag auf ein Aussetzen der Patentrechte für Covid-19 Produkte (Waiver) bei der Welthandelsorganisation (WTO). Während weit über 100 Staaten den Waiver unterstützen, blockieren einige Industrieländer. „Eine politisch erwirkte Freigabe der Patente halte ich für den falschen Weg“, sagte Bundeskanzlerin Merkel in ihrer Regierungserklärung vom 24.6.2021.[4]
WTO vertagt sich wegen Covid
"Das Vorenthalten von Impfstoffen ist im Grunde eine Fortsetzung des Kolonialismus mit anderen Mitteln." - Jörg Schaaber
Eigentlich sollte vom 30.11.-3.12.2021 die WTO in Genf tagen und eine Lösung für die Blockade beim Waiver finden. Wegen der neuen Corona-Reisebeschränkungen wurde das Treffen jedoch wenige Tage vor Beginn auf unbestimmte Zeit verschoben. Der politische Schaden ist groß. Denn mit der Vertagung rückt eine schnelle Lösung in weite Ferne, obwohl inzwischen auch die kleine aber mächtige Front der Waiver Gegner bröckelt. Sogar die USA können sich für einen Waiver erwärmen. Präsident Biden betonte kürzlich erneut, dass er sich dafür einsetzt: „Die Nachrichten über diese neue Variante [Omikron] macht es deutlicher denn je zuvor, warum die Pandemie nicht enden wird, bevor wir weltweit Impfungen haben.“[5]
Die Europäische Union setzt dagegen weiter auf Scheinlösungen. Kürzlich verkündete Handelskommissar Valdis Dombrovskis: „In den vergangenen Monaten hat sich die Europäische Union, in diesem Fall die EU-Kommission, innerhalb der WHO sehr engagiert, um zu diskutieren, wie wir Zwangslizenzen handhaben könnten.“[6] Dabei gehören Zwangslizenzen längst zum von der WTO erlaubten Instrumentarium, um Medikamente und Impfstoffe auch ohne Zustimmung des Patentinhabers produzieren zu können. Nur ist das Verfahren zeitraubend und unflexibel und deshalb in der aktuellen Krise untauglich.
Mary Robinson, ehemalige Präsidentin von Irland und UN-Menschrechtskommissarin, appelliert an die EU, die Blockade aufzugeben. Eine besondere Botschaft hat sie an Deutschland: „Deutschland, das mit der Förderung und Unterstützung des Biontech-Pfizer Impfstoffs der Welt einen enormen Dienst erwiesen hat, muss seine Schlüsselrolle spielen, indem es sich anderen europäischen Ländern anschließt, um ihn [den Waiver] Wirklichkeit werden zu lassen. Ich hoffe, dass die Regierung des kommenden Kanzlers Scholz diese frühe Gelegenheit, globale Führungsstärke zu zeigen, zu nutzen weiß.“[7] (JS)
 Der Zugang zu Covid-19-Impfungen ist global ungerecht verteilt. In den meisten afrikanischen Staaten liegt die Quote der voll Geimpften unter 10%, Schlusslicht ist die Demokratische Republik Kongo mit 0,06%.[8] In Deutschland sind dagegen 68,6% voll geimpft und 12,5% haben eine zusätzliche Boosterimpfung erhalten.[9]
Der Zugang zu Covid-19-Impfungen ist global ungerecht verteilt. In den meisten afrikanischen Staaten liegt die Quote der voll Geimpften unter 10%, Schlusslicht ist die Demokratische Republik Kongo mit 0,06%.[8] In Deutschland sind dagegen 68,6% voll geimpft und 12,5% haben eine zusätzliche Boosterimpfung erhalten.[9]
Artikel aus dem Pharma-Brief 10/2021, S.1
Bild Weltkarte Zugang Covid-19-Impfungen © WHO, Datenstand 30.11.2021
[1] www.14dd5266c70789bdc806364df4586335-gdprlock/watch?v=dD8McADeWvs [Zugriff 1.12.2021]
[2] Oxfam (2021) Profit vor Weltgesundheit: 1000 Dollar Gewinn pro Sekunde für Pharmafirmen – aber kaum Impfstoff für einkommensschwache Länder. Pressemitteilung 16. Nov. www.oxfam.de/presse/pressemitteilungen/2021-11-16-profit-weltgesundheit-1000-dollar-gewinn-pro-sekunde-pfizer [Zugriff 1.12.2021]
[3] Pharma-Brief (2021) Molnupiravir: Öffentlich entdeckt – privat kassiert? Nr. 8-9, S. 4
[4] Tagesschau (2021) Regierungserklärung vor EU-Gipfel: Merkel verteidigt Patentschutz für Impfstoffe. 24. Juni www.tagesschau.de/inland/merkel-regierungserklaerung-155.html
[5] Reuters (2021) U.S. President Biden calls for intellectual property protection waivers after Omicron discovery. 27 Nov www.reuters.com/business/healthcare-pharmaceuticals/us-president-biden-calls-intellectual-property-protection-waivers-covid-19-2021-11-26
[6] Reiche M (2021) Streit über Patente: Zwangslizenzen für Corona-Impfstoff? Tagesschau 1.12.2021 www.tagesschau.de/wirtschaft/unternehmen/patente-freigabe-corona-impstoff-101.html [Zugriff 1.12.2021]
[7] Robinson M (2021) Vaccine waiver is a moment of truth for EU values. Politico 1 Dec www.politico.eu/article/vaccine-waiver-eu-values-coronavirus
[8] WHO (2021) WHO Coronavirus dashboard. Datenstand 30.11.2021 https://covid19.who.int/ [Zugriff 1.12.2021]
[9] RKI (2021) Tabelle mit den gemeldeten Impfungen nach Bundesländern und Impfquoten nach Altersgruppen. Datenstand 1.12.2021 www.rki.de/DE/Content/InfAZ/N/Neuartiges_Coronavirus/Daten/Impfquotenmonitoring.xlsx
Covid-19: Drei Dosen oder keine
Impfstoff-Zugang bleibt global ungerecht
Während in großen Teilen der Welt die meisten Menschen noch keine Covid-19-Impfung erhalten haben, beginnen Industrieländer eine dritte Dosis zu impfen.
In Europa wurden pro 100 EinwohnerInnen inzwischen 101 Dosen verimpft, auf dem afrikanischen Kontinent gerade einmal 8,4.[1] Entgegen den Empfehlungen der Weltgesundheitsorganisation haben etliche Industrieländer damit angefangen, eine dritte Dosis zu impfen.[2] Das ist nicht nur krass ungerecht und kostet Menschenleben. Es ist auch epidemiologisch unsinnig. Denn solange Covid-19 weiter grassiert, bilden sich immer neue Varianten und Viren kennen bekanntlich keine Grenzen.
Der Waiver für Covid-19-Patente bei der WTO wird von Industrieländern weiter blockiert. Damit verschieben sich auch die Aussichten auf eine massive globale Ausweitung der Impfstoffproduktion immer weiter in die Zukunft. Darüber können auch einige von den Patentinhabern kontrollierte neue Produktionsstätten im globalen Süden nicht hinwegtäuschen. So die Kooperation von Biovac (Südafrika) mit Biontech/Pfizer, die ab 2022 rund 100 Millionen Dosen für Afrika produzieren soll.[3] Die Preiskonditionen sind dabei unbekannt.
Covax bleibt Trostpflaster
Das internationale Public Private Partnership Covax kann die Lücke auch nicht füllen und bleibt sogar hinter seinen eigenen bescheidenen Ansprüchen zurück. Bis Ende August wurden weltweit gerade einmal 230 Millionen Dosen ausgeliefert, die sich auf 139 Länder verteilen.[4]
Deutschland hat zwar 100 Millionen Dosen an Covax versprochen, die „hauptsächlich an Entwicklungsländer“ gehen sollen.[5] Ausgeliefert werden davon bis Mitte September aber nur 1,3 Millionen Dosen. Zum Vergleich: In Deutschland wurden bis zum 9. September 103,8 Millionen Dosen verimpft, damit sind 72,6% der Bevölkerung ab 18 Jahren vollständig geschützt.[6] Das Abgeben an Covax fällt nicht besonders schwer, denn insgesamt hat sich Deutschland für 2021 viel mehr gesichert, als gebraucht wird: 313 Millionen Dosen.[7]
Von Süd nach Nord
Besonders bizarres Beispiel der globalen Ungerechtigkeit: Die südafrikanische Firma Aspen füllt bereits den Johnson & Johnson-Impfstoff ab. Der Vertrag wurde schon im November 2020 geschlossen.[8] Südafrika hatte mit Johnson & Johnson eine Lieferung von 31 Millionen Dosen für 2021 vereinbart.[9] Davon hat das Land bis Ende Juni gerade einmal 1,5 Millionen erhalten. In den letzten Monaten wurden aber mindestens 32 Millionen Dosen aus Südafrika exportiert, schwerpunktmäßig nach Europa, unter anderem auch nach Deutschland und Spanien.
Das war möglich, weil Südafrika einen Vertrag unterschreiben musste, der Exportbeschränkungen untersagte. Pop Maja, Sprecher des Gesundheitsministerium, bedauerte: „Die Regierung hatte keine Wahl – unterzeichne den Vertrag oder es gibt keinen Impfstoff.“5 Darüber hinaus enthält die Vereinbarung mit Südafrika auch noch andere fragwürdige Klauseln. So können nicht nur Betroffene keine Impfschäden gegen Johnson & Johnson geltend machen, auch eine Mängelhaftung des Herstellers gegenüber der südafrikanischen Regierung ist ausgeschlossen.
Seit Juli füllt Aspen nun 60% der Dosen für Afrika und 40% für Europa ab. Produziert wird derzeit mit Wirkstoff aus den Niederlanden. Die günstigere Verteilung beruht auf einem Entgegenkommen der Europäischen Union, die angesichts der Krise jetzt darauf verzichtet, wie ursprünglich mit Johnson & Johnson vereinbart, nur 10% der Dosen in Afrika zu lassen.
Erst nach heftigen Protesten machte die EU eine Kehrtwende und sagte am 2. September zu, dass alle bereits exportierten Dosen zurückgeschickt werden und künftig die gesamte südafrikanische Produktion von Aspen in Afrika bleibt.[10]
Dass in Südafrika inzwischen 10% der Bevölkerung voll geimpft ist,4 ist hauptsächlich Bestellungen von Biontech-Impfstoff durch die Regierung und eine Spende von 5,7 Millionen Dosen durch die USA zu verdanken.[11] Der Todeszoll bleibt wegen der geringen Impfrate hoch: Anfang September starben innerhalb einer Woche 1.824 SüdafrikanerInnen mit Covid-19.[12]
Schlechter? Preis erhöhen!
Obwohl sich zeigt, dass die Wirkung der bislang zugelassenen Impfstoffe gegen die Delta-Variante geringer ist, haben die wichtigsten Lieferanten für die EU die Preise erhöht, wie Anfang August bekannt wurde.[13] Biontech/Pfizer verlangt nun 26% mehr,[14] Moderna 13%.[15] Angesichts einer möglicherweise notwendigen dritten Dosis nimmt Biontech/Pfizer dann 63% mehr ein. Wahrhaft ein gutes Geschäft. Vorbei die Zeiten, in denen Produkte, die ihre Versprechen nicht hielten, billiger wurden. (JS)
Biontech Rekordumsatz
Nach einem Rückgang im vergangenen Jahr wächst das Bruttosozialprodukt in Deutschland dieses Jahr voraussichtlich um 4%. Allein das Umsatzwachstum von Biontech trägt dazu 0,5% bei. Die Firma erwartet 2021 mit ihrem Covid-19 Impfstoff einen Umsatz von 15,9 Mrd. €. Sebastian Dullien, Direktor des Instituts für Makroökonomie und Konjunkturforschung (IMK), sagte dazu, dass ihm kein anderer Fall bekannt ist, wo eine einzelne Firma einen so starken Einfluss auf das Wachstum gehabt hat.[16]
Artikel aus dem Pharma-Brief 7/2021, S.6
[1]Holder J (2021) Tracking Coronavirus Vaccinations Around the World. 7 Sept. www.nytimes.com/interactive/2021/world/covid-vaccinations-tracker.html [Zugriff 7.9.2021]
[2]Reuters (2021) Factbox-Countries weigh need for booster COVID-19 shots. 24 Aug. www.reuters.com/article/us-health-coronavirus-booster-idUKKBN2FP168
[3]Russell R (2021) Battling vaccine inequity: Africa boosts Covid-19 jab production capacity. Standard 17 Aug. www.standard.co.uk/optimist/vaccine-world/covid-vaccine-inequity-africa-production-capacity-b951012.html
[4]GAVI (2021) COVAX Global Supply Forecast. 8 Sept. www.gavi.org/news/document-library/covax-global-supply-forecast
[5]Gavi (2021) First delivery of German-donated COVID vaccines to COVAX land in Mauritania, with other deliveries to follow. Press release 8 Sept. www.gavi.org/news/media-room/first-delivery-german-donated-covid-vaccines-covax-land-mauritania-other-deliveries
[6]RKI (2021) Impfquotenmonitoring. Datenstand 9. Sept. www.rki.de/DE/Content/InfAZ/N/Neuartiges_Coronavirus/Daten/Impfquotenmonitoring.xlsx?__blob=publicationFile [Zugriff 10.9.2021]
[7]BMG (2021) Lieferprognosen der verschiedenen Hersteller für das Jahr 2021 (Stand 22.06.2021) www.bundesgesundheitsministerium.de/fileadmin/Dateien/3_Downloads/C/Coronavirus/Impfstoff/Lieferprognosen_verschiedene_Hersteller_2021.pdf [Zugriff 10.9.2021]
[8]Aspen (2020) Aspen announces agreement with Johnson & Johnson to manufacture investigational COVID-19 vaccine candidate. 2 Nov. www.aspenpharma.com/2020/11/02/aspen-announces-agreement-with-johnson-johnson-to-manufacture-investigational-covid-19-vaccine-candidate
[9]Robbins R and Mueller B (2021) Covid Vaccines Produced in Africa Are Being Exported to Europe. New York Times 16 Aug. www.nytimes.com/live/2021/08/16/world/covid-delta-variant-vaccine#while-south-africa-waits-for-vaccine-supplies-jj-doses-made-there-are-sent-to-europe
[10]Jerving S (2021) Deal to send COVID-19 vaccines from South Africa to Europe dismantled. DEVEX 2 Sept. www.devex.com/news/deal-to-send-covid-19-vaccines-from-south-africa-to-europe-dismantled-101532
[11]US Embassy in South Africa (2021) United States Donates 5.7 Million COVID-19 Doses to South Africa. Press release 28 July https://za.usembassy.gov/united-states-donates-5-7-million-covid-19-doses-to-south-africa/
[12]WHO (2021) Covid-19 dashboard https://covid19.who.int/region/afro/country/za [Abruf 7.9.2021]
[13]Reuters (2021) Pfizer and Moderna raise prices for COVID-19 vaccines in EU- FT. 2 Aug. www.reuters.com/business/healthcare-pharmaceuticals/pfizer-moderna-raises-prices-its-covid-19-vaccines-eu-ft-2021-08-01
[14]19,50 € statt bislang 15,50 €
[15]21,48 € (25,50 US$) statt 19,00 €
[16]Reuters (2021) BioNTech alone could lift German economy by 0.5% this year. 10. Aug. www.reuters.com/article/us-germany-economy-biontech-idUSKBN2FB15A
Covid-19, trust, and Wellcome
Wie sich die Investitionen der Stiftung in die Pharmabranche mit ihren eigenen Forschungsanstrengungen überlappen
Der Hauptgeldgeber der Gesundheitsforschung profitiert finanziell von der Pandemie; das wirft Fragen zu Transparenz und Verantwortlichkeit auf. Ein Beitrag aus dem BMJ von Tim Schwab.*
Ein Merkmal der Covid-19-Pandemie wird immer deutlicher: Die Antwort des öffentlichen Gesundheitswesens wird nicht nur von Regierungen und multilateralen Institutionen wie der Weltgesundheitsorganisation (WHO) vorangetrieben, sondern auch von einer Flut von Öffentlich-Privaten Partnerschaften, bei denen Arzneimittelfirmen und private Stiftungen mitwirken.
Eine neue führende Stimme ist dabei der Wellcome Trust, einer der weltgrößten Geldgeber für Gesundheitsforschung, zu seinen sich immer weiter ausbreitenden wohltätigen Aktivitäten in der Pandemie gehört es, gemeinsam mit der WHO ein Programm zur Unterstützung neuer Covid-19-Therapien zu leiten. Das Projekt “Access to Covid-19 Tools (ACT) Accelerator” hofft, Milliarden Dollar einzuwerben und im laufenden Jahr hunderte Millionen von Behandlungskursen zur Verfügung zu stellen, darunter Dexamethason und einer Reihe von monoklonalen Antikörpern.[1]
Das BMJ fand heraus, dass Wellcome gleichzeitig Beteiligungen an Firmen hält, die genau diese Behandlungen herstellen. Finanzmitteilungen von Ende 2020 zeigen, dass Wellcome mit 275 Mio. £ (318 Mio. €; 389 Mio. US$) an Novartis beteiligt ist. Die Firma stellt Dexamethason her und erforscht weitere Therapien. Und Roche, an der Wellcome mit 252 Mio £ beteiligt ist,[2] hilft dem Biotechnologie-Unternehmen Regeneron bei der Herstellung von monoklonalen Antikörpern. Beide – Roche und Novartis – berichten, dass sie mit dem WHO “ACT Accelerator” Gespräche über ihre therapeutischen Arzneimittel geführt hatten.[3]
Die finanziellen Interessen von Wellcome wurden auf der Webseite des Trusts veröffentlicht und sind durch gesetzlich geforderte Angaben bei den Finanzbehörden öffentlich zugänglich. Sie sind aber anscheinend nicht als finanzielle Interessenskonflikte im Zusammenhang mit der Arbeit von Wellcome zu Covid-19 offengelegt worden, obwohl sie zeigen, dass der Trust potenziell finanziell von der Pandemie profitieren kann.
Auf Enthüllungen der finanziellen Interessenskonflikte des Wellcome Trusts folgen auf Berichte über eine andere Wohltätigkeitsorganisation, die Gates Stiftung, die ebenso in der Lage ist, aus ihrer führenden Rolle bei der Pandemiebekämpfung finanziellen Nutzen zu ziehen. Eine Recherche von The Nation enthüllte, dass Gates mehr als 250 Mio. US$ (179 Mio. £; 206 Mio. €) in Firmen investiert hatte, die zu Covid-19 arbeiten, und zitierte zivilgesellschaftliche Gruppen, die über den übergroßen Einfluss alarmiert waren, den die milliardenschwere Wohltätigkeitsorganisation in der Pandemiebekämpfung ausübt; sie sehen das als eine Stärkung der Rolle der Pharmaindustrie.[4]
Doch Stiftungen wie die von Gates und Wellcome — und sogar Pharmakonzerne — werden während der Pandemie im Allgemeinen in den Medien für ihre Anstrengungen gepriesen, die Gesundheitskrise zu lösen. Ihre finanziellen Interessen erfahren dagegen relativ wenig Aufmerksamkeit und ihre Arbeit ist wenig externer Kontrolle unterworfen.
„Die Pandemie trägt gerade dazu bei, den Ruf von Organisationen wie Gates und Wellcome und der Pharmakonzerne aufzupolieren. Ich glaube nicht, dass sie das wirklich verdienen”, sagt Joel Lexchin, emeritierter Professor der School of Health Policy and Management der York University in Toronto. „Ich denke, sie handeln, wie sie es immer getan haben. Das heißt aus Sicht der Pharmafirmen, ihre eigenen finanziellen Interessen zu verfolgen, und aus der Sicht der Stiftungen, ihre eigenen privat entwickelten Ziele zu verfolgen, ohne irgendjemand außer ihren eigenen Vorständen verantwortlich zu sein.“
Interessenkonflikte?
Mohga Kamal-Yanni, eine Politikberaterin von UNAIDS und anderen Organisationen, arbeitete vor kurzem an einem Paper mit, das auf Probleme mit dem Einfluss der Gates Stiftung in der Pandemie hinwies. Sie sagt, dass die Investitionen des Wellcome Trusts kritische Fragen rund um das Thema Transparenz und Verantwortlichkeit aufwerfen.[5]
„Bei Covid haben diese beide Worte eine so riesige Bedeutung, weil wir wissen müssen, ob Entscheidungen auf der Grundlage von Evidenz und Wissenschaft getroffen werden,“ sagt sie zum BMJ. „Wissen wir, mit welchen Firmen sie sprechen? Wie sie entscheiden, welche Firma sie finanziell fördern? — oder dieses oder ein anderes Produkt?“
Der Wellcome Trust bestreitet, dass seine Investitionen seine Unabhängigkeit kompromittieren — oder gar in Widerspruch zu ihr geraten. „Wir sind uns keiner Situation in unseren Beziehungen mit … dem ACT Accelerator bewusst, in der als Folge unseres Investmentportfolios ein Konflikt entstanden ist oder in der es für Repräsentanten von Wellcome notwendig gewesen wäre, sich wegen Befangenheit zurückzuziehen,“ sagte ein Sprecher und lehnte es ab, seine Investitionen in Novartis oder Roche zu kommentieren. „Wir würden niemals Entscheidungen zur Pandemiebekämpfung treffen oder Dritten Empfehlungen aus anderen Gründen als der Sorge um die öffentliche Gesundheit geben.“
Unterstützer von Wellcome beschreiben die sehr kompetente biomedizinische Expertise, die die Stiftung in der Pandemie einbringt, in herausragender Weise mit seinem Direktor, Jeremy Farrar, einem berühmten Erforscher von Infektionskrankheiten, dem der Verdienst zugeschrieben wird, bei früheren Ausbrüchen von Ebola und Vogelgrippe eine führende Rolle gespielt zu haben.[6][7]
Kenny Baillie, Leiter einer Forschungsgruppe in der Abteilung Genetics and Genomics an der University of Edinburgh, der Forschungsgelder von Wellcome erhalten hat, sagt, dass die Stiftung Anerkennung verdient als ein “Leuchtturm von Integrität und verantwortungsvoller Unternehmensführung.”
Er erklärt: „Ich kann sicherlich aus eigener persönlicher Erfahrung in der Zusammenarbeit auf der Wissenschaft sprechen, da gab es keinen Versuch, mich oder andere ForscherInnen, die ich kenne, zu beeinflussen und abzuhalten, die beste Wissenschaft zum Wohl der Menschen zu liefern.“ Doch es bleibt unklar, welche Governance-Strukturen bestehen, die garantieren, dass Wellcomes riesiges Stiftungsvermögen das Agenda-Setting bei der WHO oder andere Arbeit der Stiftung in der Pandemie nicht beeinflusst.
Unitaid, das mit federführend für das WHO ACT Accelerator Project ist, sagt, dass es mit Wellcome ein „klares gegenseitiges Einvernehmen gibt, dass relevante institutionelle Interessen transparent offengelegt werden.“ Aber Unitaid teilte dem BMJ vergangenen Dezember mit: “Wir haben noch keine einzige Erklärung zu Interessenkonflikten erhalten.“
Marc Rodwin, Rechtsprofessor an der Suffolk University in Boston, Massachusetts, sagt, dass Institutionen mit finanziellen Interessenskonflikten dennoch wertvolle Beiträge zur Pandemiebekämpfung leisten könnten, aber keine Einfluss- oder Entscheidungsposition haben sollten.
„Ich würde sogar noch weiter gehen, als nur zu sagen, sie sollten sich vor bestimmten Entscheidungen als befangen erklären. Warum werden sie überhaupt ausgewählt für diese Stellen mit Machtbefugnissen?“, fragt er. „Ich mag hier das Konzept des epidemiologischen Risikofaktors – es führt nur ein Risikolevel ein, das unnötig ist. Wenn viel Geld im Spiel ist, willst du nicht solche finanziellen Konflikte haben, die diese Entscheidungen beeinflussen.“
Regierungsberater
Zusätzlich zur Arbeit durch die WHO beeinflusst Wellcome die Pandemiebekämpfung auch durch Farrars Stellung in der Scientific Advisory Group for Emergencies, die die Regierung des Vereinigten Königreichs zu Covid-19 berät; er hat zudem einen Sitz im Vorstand der Coalition for Epidemic Preparedness Innovations, einer führenden Öffentlich-Privaten Partnerschaft in der Pandemie, die mehr als eine Mrd. US$ für die Entwicklung von Covid-19-Impfstoffen zugesagt hat.[8] Auch tritt Farrar häufig als Experte in den Medien auf, einschließlich dem BMJ, wo er das Potenzial bestimmter Medikamente gegen Covid-19 hervorhob.[9],[10] Diese Berater- und Medienaktivitäten weisen Überschneidungen zu den 28 Mrd. £ Stiftungskapital des Wellcome Trust auf, der mindestens 1,25 Mrd. £ in Firmen investiert hat, die zu Covid-19-Impfstoffen, Therapeutika und Diagnostika arbeiten: Roche, Novartis, Abbott, Siemens, Johnson & Johnson, und — durch Beteiligungen an der Investmentgesellschaft Berkshire Hathaway — Merck, AbbVie, Biogen, and Teva.[11]
Farrar sitzt in dem internen Investment-Ausschuss von Wellcome, der in Bezug auf das Stiftungskapital eine wichtige beratende Rolle spielt.[12] Wellcome will sich nicht dazu äußern, ob die Doppelrolle Farrars wünschenswert ist — Hilfe beim Management des Stiftungsvermögens und die karitative Mission —, und lehnte mehrfach Nachfragen nach einem Interview mit Farrar oder anderen VertreterInnen der Stiftung ab.
Das ganze Ausmaß der Investitionen von Wellcome in Firmen, die zu Covid-19 arbeiten, ist nicht bekannt, da die Stiftung sich auch weigerte, gegenüber dem BMJ Details zu ihrem Investmentportfolio offenzulegen, das meiste davon wird nur allgemein beschrieben als Investition in „Hedge-Fonds“, „Aktien“, „Firmenbeteiligungen “ oder „Immobilien“.[13] Wellcome veröffentlicht auf seiner Webseite eine Liste von einigen Dutzend seiner größten individuellen Unternehmensinvestitionen. Und das BMJ enthüllte zusätzliche Informationen über Wellcomes Stiftungsvermögen durch Einsicht in Unterlagen, die die Stiftung bei der US-Finanzaufsicht einreichte.
Auf der Basis dieser begrenzten öffentlichen Berichterstattung scheint es, als expandiere Wellcome seine Covid-bezogenen Investitionen. Die Stiftung berichtete letzten Sommer den Besitz von fast zwei Millionen Aktien von Abbott Laboratories, einem Hauptlieferant von diagnostischen Tests für Covid-19.[14] Den von Wellcome eingereichten Finanzunterlagen kann entnommen werden, dass der Wert seiner 1,95 Millionen Anteile an Abbott zwischen Juli und Oktober 2020 von 178 Mio. US$ auf 212 Mio. US$ angewachsen ist, ein unerwarteter Gewinn für die gemeinnützige Stiftung.[15]
Wellcome berichtet von 3.3 Mrd. £ Gewinn aus allen Investitionen im Jahr 2020, dreimal so viel Geld, wie der Trust für wohltätige Zwecke ausgeschüttet hat.
Der Einfluss von Investoren
Die Rolle von Wellcome auf den Finanzmärkten während der Pandemie hat sich auch auf andere markante Weise niedergeschlagen. Das Wall Street Journal hat berichtet, dass Wellcome schon im Januar 2020 Telefonkonferenzen mit privaten Investmentfirmen abgehalten hat, wo Farrar Vermögensverwalter vor dem Ernst von Covid-19 warnte.[16] Die Gespräche trieben die Investoren an, ihre Portfolios zu reorganisieren, entweder um Verluste zu minimieren oder finanzielle Gewinne zu erzielen, so die Zeitung.
Die Stiftung war nicht bereit, Protokolle von Farrars Telefonkonferenz mit fremden Kapitalgebern zu liefern, und erklärte stattdessen, dass er Investoren die gleichen Covid-Warnungen anbiete wie den Nachrichtenmedien und bei anderen Treffen.
Zwei Investmentgesellschaften — Sequoia und Blackstone—, die an den Gesprächen mit Farrar teilgenommen hatten, zahlten in den letzten Jahren Dividenden an Wellcome aus, wie die Steuerveranlagung der Stiftung in den USA kürzlich zeigte. Wellcome wollte nicht kommentieren, ob sie zu dem Zeitpunkt Geld in die Firmen investiert hatte, als die Stiftung Telefongespräche von Farrar mit eben diesen Unternehmen organisiert hatte.[17]
Die Ethik der Investmentaktivitäten von Wellcome wurde in den vergangenen Jahren mehrfach öffentlich in Frage gestellt. Unter anderem in einer öffentlichen Kampagne, die 2015 vom Guardian organisiert wurde, um auf Wellcome und die Gates Stiftung Druck auszuüben, sich von Investitionen in fossile Brennstoffe zu trennen. Zehntausende unterzeichneten eine Petition, die argumentierte, dass die Investments von Wellcome und Gates in fossile Brennstoffe im Widerspruch zu ihrer Arbeit zur Förderung des „menschlichen Fortschritts und der Gleichheit“ stehe.[18]
Farrar lehnte in einem Antwortschreiben Desinvestition (Divestment) als Strategie ab. Er sagte, Wellcome nutze seine Stellung als Investor dazu, Firmen fossiler Brennstoffe zu einem besseren Verhalten zu drängen.[19] Ein früherer Mitarbeiter von Wellcome berichtete dem BMJ, dass das Investment der Stiftung in fossile Brennstoffe zu vielen Auseinandersetzungen unter den Mitarbeitern führte, die diese Investmentstrategie der Stiftung in Frage stellten.
Die Zeitschrift Science berichtete 2018, dass Wellcome fast 1 Mrd. US$ an Offshore-Investitionen hielt, einschließlich eines Cayman Islands Energiefonds, der Anteile an einer Firma hielt, die extrem umweltbelastende Schiffstreibstoffe verkauft. Science zitierte Ökonomen, die Offshore-Investitionen von Wellcome und anderen Stiftungen in Steueroasen wie den Cayman Islands scharf kritisierten und sie beschuldigten, ein Verhalten der Steuervermeidung zu institutionalisieren und zu normalisieren, was die Einkommensungleichheit verschärft.[20]
Governance des öffentlichen und privaten Sektors
Während der ganzen Pandemie kursierten Vorwürfe finanzieller Interessenskonflikte öffentlicher und privater Akteure in vielen Ländern. Im Vereinigten Königreich geriet der wissenschaftliche Chefberater der Regierung, Patrick Vallance, in die Schlagzeilen, als ihm nachgewiesen wurde, finanzielle Verbindungen zu der Pharmafirma GlaxoSmithKline zu haben.[21] Regierungsquellen haben Vallance gegen Vorwürfe von Fehlverhalten verteidigt.
In den USA wurden gegen vier Kongressmitglieder Untersuchungen eingeleitet, weil sie Aktienhandel mit nicht-öffentlichen Informationen betrieben, zu denen sie auf Grund ihrer politischen Stellung Zugang hatten. Alle wurden sie während der Untersuchung freigesprochen, berichtete die New York Times.[22],[23]
Im letzten Jahr berichtete das BMJ über ein Versagen der Scientific Advisory Group for Emergencies des Vereinigten Köngreichs, konkurrierende Interessen in Bezug auf Covid-19 zu veröffentlichen. Daraufhin wurden sie zur öffentlichen Überprüfung freigegeben.[24]
Trotz der herausragenden Rolle, die private Wohltätigkeitsorganisationen in der Pandemiebekämpfung spielen, wurden ihre finanziellen Interessen wenig hinterfragt, wohl weil Stiftungen nicht den gleichen Aufsichtsmechanismen unterworfen sind wie öffentliche Institutionen.
Linsey McGoey, eine Soziologieprofessorin an der University of Essex, die extensiv über Verantwortlichkeit in der Philanthropie geschrieben hat, sieht einen Zusammenhang zwischen den Investment von Wellcome und Gates in die Pharmaindustrie im und ihrer Unterstützung für die herrschenden Marktmechanismen, die die moderne Medizin antreiben – was dazu führt, dass die reichen Länder bevorzugten Zugang zu Covid-19 Medikamenten bekommen.[25] Viele Stakeholder stellen dieses ökonomische Modell während der Pandemie in Frage, stellt McGoey fest, und sie üben Druck auf die Welthandelsorganisation aus, Einschränkungen durch geistige Eigentumsrechte in Bezug auf Impfstoffe und Therapeutika zu lockern.[26]
Sie sagt, „Sie scheinen sich ganz einem gemeinnützigen Modell verpflichtet zu fühlen …[das] in Wirklichkeit aber dem Ansatz von Gesundheits- und Impfstoffgerechtigkeit, den die meisten AktivistInnen und politischen EntscheidungsträgerInnen des globalen Südens fordern, zu widersprechen scheint.“
„Diese Art von Stiftungen erhalten den falschen ideologischen Eindruck aufrecht, dass sie [...] das Problem lösen, auch wenn sie gerade das nicht tun. Und sie könnten es noch verstärken, indem sie den ideologischen Eindruck perpetuieren, der privaten Sektor nehme die Rolle des Retters ein.“
Tim Schwab, Freier Journalist Diese E-Mail-Adresse ist vor Spambots geschützt! Zur Anzeige muss JavaScript eingeschaltet sein!
*Dieser Artikel erschien am 3.3.2021 im BMJ; 372, p n556 https://doi.org/10.1136/bmj.n556 . Wir danken dem BMJ für die Abdruckgenehmigung. Übersetzung: Margit Urhahn
Interessenkonflikte: Tim Schwab hat die Politik des BMJ zu Interessenkonflikten gelesen und verstanden. Er erklärt, dass er Empfänger eines Alicia Patterson Foundation journalism fellowship 2019 ist. Das BMJ veröffentlicht Arbeiten von Forschungsgruppen und AkedemikerInnen, die von der Bill and Melinda Gates Foundation und dem Wellcome Trust gefördert werden.
Hinweis für den englischsprachigen Originalartikel: This article is made freely available for use in accordance with BMJ‘s website terms and conditions for the duration of the covid-19 pandemic or until otherwise determined by BMJ. You may use, download and print the article for any lawful, non-commercial purpose (including text and data mining) provided that all copyright notices and trade marks are retained. https://bmj.com/coronavirus/usage
Artikel aus dem Pharma-Brief 3-4/2021, S.4
Wir behalten im Folgenden die Zitierweise des BMJ bei:
[1] Schreier P. A look inside the global partnership that’s working to find and deliver Covid-19 treatments. Wellcome Trust. 29 Oct 2020. https://wellcome.org/news/look-inside-global-partnership-thats-working-find-and-deliver-covid-19-treatments
[2] Wellcome Trust. List of directly held equity holdings as of September 2020. https://wellcome.org/about-us/investments/direct-public-equity-holdings
[3] Guarascio F. Exclusive: WHO-led COVID drug scheme doubles down on antibodies, steroids and shuns remdesivir. Reuters 2020 Nov 5. https://www.reuters.com/article/us-health-coronavirus-who-drugs-exclusiv/exclusive-who-led-covid-drug-scheme-doubles-down-on-antibodies-steroids-and-shuns-remdesivir-idUSKBN27L1OY
[4] Schwab T. While the poor get sick, Bill Gates just gets richer. Nation 2020 Oct 5. https://www.thenation.com/article/economy/bill-gates-investments-covid/
[5] Malpani R, Baker B, Kamal-Yanni M. Corporate charity—is the Gates Foundation addressing or reinforcing systemic problems raised by covid-19? Health Policy Watch 2020 Oct 31. https://healthpolicy-watch.news/gates-foundation-address-systemic-covid-19/
[6] Rosenthal E. On the front: a pandemic is worrisome but unlikely. New York Times 2006 Mar 28. https://www.nytimes.com/2006/03/28/health/on-the-front-a-pandemic-is-worrisome-but-unlikely.html
[7] McKie R. “We can beat Ebola but must prepare for what comes next,” says Wellcome Trust head. Guardian 2019 Dec 22. https://www.theguardian.com/world/2019/dec/22/we-can-beat-ebola-prepare-for-health-battles-to-come-jeremy-farrar-wellcome-trust
[8] UK Government Office of Science. List of participants of SAGE and related sub-groups. https://www.gov.uk/government/publications/scientific-advisory-group-for-emergencies-sage-coronavirus-covid-19-response-membership/list-of-participants-of-sage-and-related-sub-groups. (CEPI list of board members and COVID vaccine support is available at www.cepi.net.)
[9] Farrar J. Soon Covid-19 will be treatable, but it shouldn’t just be the rich who benefit. Guardian 2020 Oct 12. https://www.theguardian.com/commentisfree/2020/oct/12/covid-19-treatable-vaccines-treatments
[10] Looi M-K. Jeremy Farrar: Make vaccine available to other countries as soon as our most vulnerable people have received it. BMJ2021;372:n459. doi:10.1136/bmj.n459 pmid:33608412
[11] Wellcome Trust. Annual report and financial statement 2020; list of directly held equity holdings as of September 2020. https://wellcome.org/about-us/investments/direct-public-equity-holdings
[12] Wellcome Trust. Investment committee list. https://wellcome.org/about-us/investments. (Investment policy, 2020 Feb 24, is available at https://wellcome.org/sites/default/files/investment-policy-202002.pdf.)
[13] Wellcome Trust. Annual report and financial statement 2020. https://wellcome.org/reports/wellcome-annual-report-2020
[14] Abbott Laboratories. US Securities and Exchange Commission 10Q regulatory filing for the quarterly period ended September 30, 2020. https://www.abbottinvestor.com/node/32711/html
[15] Wellcome Trust. US Securities and Exchange Commission 13F regulatory filings. 10 Jul and 20 Oct 2020. https://www.sec.gov/Archives/edgar/data/1026720/000101297520000777/xslForm13F_X01/infotable.xml
[16] Chung J. The early coronavirus warning that woke up Wall Street. Wall Street J 2020 Jun 12. https://www.wsj.com/articles/the-early-coronavirus-warning-that-woke-up-wall-street-11591954202
[17] Wellcome Trust. Annual 990 tax filings with the United States Internal Revenue Service.
[18] Howard E. Keep it in the ground campaign: six things we’ve learned. Guardian 2015 Mar 25. https://www.theguardian.com/environment/keep-it-in-the-ground-blog/2015/mar/25/keep-it-in-the-ground-campaign-six-things-weve-learned. The Guardian’s “Keep it in the Ground” petition can be found at https://www.theguardian.com/environment/ng-interactive/2015/mar/16/keep-it-in-the-ground-guardian-climate-change-campaign
[19] Farrar J. Fossil fuel investments are an issue on which fair-minded people will disagree. Guardian 2015 Apr 17. https://www.theguardian.com/environment/keep-it-in-the-ground-blog/2015/apr/17/fossil-fuel-investments-are-an-issue-on-which-fair-minded-people-will-disagree
[20] Piller C. Private research funders court controversy with billions in secretive investments. Science 2018 Dec 6. https://www.sciencemag.org/news/2018/12/private-research-funders-court-controversy-billions-secretive-investments
[21] Thacker PD. Conflicts of interest among the UK government’s covid-19 advisers. BMJ2020;371:m4716. doi:10.1136/bmj.m4716 pmid:33298559
[22] Benner K, Fados N. Justice Dept ends inquiries into 3 senators’ stock trades. New York Times 2020 May 26. https://www.nytimes.com/2020/05/26/us/politics/senators-stock-trades-investigation.html
[23] Fandos N, Benner K. Justice Dept ends stock trade inquiry into Richard Burr without charges. New York Times. 19 Jan 2021. https://www.nytimes.com/2021/01/19/us/politics/richard-burr-stock-trades-investigation.html
[24] Coombes R. Covid-19: SAGE members’ interests published by government 10 months into pandemic. BMJ2020;371:m4911. doi:10.1136/bmj.m4911 pmid:33334782
[25] Dyer O. Covid-19: Many poor countries will see almost no vaccine next year, aid groups warn. BMJ2020;371:m4809. doi:10.1136/bmj.m4809 pmid:33310819
[26] India and South Africa ask WTO to waive rules to aid COVID-19 drug production. Reuters 2020 Oct 4. https://www.reuters.com/article/uk-health-coronavirus-wto/india-and-south-africa-ask-wto-to-waive-rules-to-aid-covid-19-drug-production-idUKKBN26P0H3?edition-redirect=uk
Covid-19 in Ghana: Basisversorgung sicherstellen
Auch Mutter-Kind-Gesundheit gefährdet
Ghana hat auf die Pandemie schnell reagiert und die Zahl der Todesfälle durch Covid-19 ist bisher verhältnismäßig niedrig. Doch die Gesundheitsversorgung hat stark gelitten. Viele Einrichtungen konnten übliche Versorgungsleistungen nicht anbieten, weil es wegen der Pandemie an Testkapazitäten mangelte und Medikamente knapp wurden. Auch Impfkampagnen wurden ausgesetzt. Mit Unterstützung der Weltgesundheitsorganisation (WHO) werden nun Konzepte umgesetzt, um grundlegende Versorgungsleistungen auch unter Pandemiebedingungen zu gewährleisten.
Rund 118.000 Infektionen und rund 1.000 Todesfälle durch Covid-19 zählte Ghana bis Ende August,[1] die meisten davon in städtischen Ballungsgebieten wie Accra und Kumasi. Bereits sehr früh hatte die Regierung Maßnahmen ergriffen, um die Ausbreitung der Pandemie einzudämmen. Die Ausstattung von Laboren wurde verbessert, Quarantäne- und Isolationsstationen eingerichtet, Krankenhauskapazitäten aufgestockt. Im Februar hatte Ghana als erster Staat der Welt Corona-Impfstoff über das Covax-Programm erhalten und damit begonnen, die Bevölkerung zu impfen.[2]
Doch die Lage im Gesundheitssystem ist durch die Pandemie äußerst angespannt: Für 1.000 Menschen steht nur ein Krankenhausbett zur Verfügung. Nicht einmal zwei ÄrztInnen und gerade einmal 42 Pflegekräfte und Hebammen kommen in Ghana auf 10.000 EinwohnerInnen.[3] Eine flächendeckende Versorgung der PatientInnen ist vor allem in ländlichen Gebieten nicht gewährleistet. Gesundheitsdienste und Präventionsarbeit wurden wegen der Pandemie eingeschränkt, Impfkampagnen ausgesetzt.
Müttergesundheit vernachlässigt
Schon vor der Pandemie war die Müttersterblichkeit in Ghana hoch: Pro 100.000 Lebendgeburten starben 319 Mütter durch Schwangerschaft oder Geburt.[4] In Deutschland sind es sieben.[5] Grund dafür ist vor allem die mangelnde Vorsorge während der Schwangerschaft: Etwa ein Drittel aller Gebärenden nimmt Vorsorgetermine nicht wahr oder hat keinen Zugang dazu. Covid-19 hat diese Situation noch verschlechtert. Agbozo und Jahn untersuchten die Auswirkungen der Pandemie auf die Versorgung von Schwangeren in der ersten Jahreshälfte 2020 und konstatieren einen starken Rückgang an Gesundheitschecks und Blutuntersuchungen in den von Covid-19 besonders betroffenen städtischen Regionen.[6] Um Kontakte zu vermeiden, wurden Vorsorgetermine in das zweite Drittel der Schwangerschaft verlegt und Untersuchungsabstände wurden vergrößert, sofern keine Risiko-Schwangerschaft vorlag. Aus Angst vor Ansteckung vermieden außerdem viele Schwangere den Besuch in Gesundheitseinrichtungen. Die größte Gesundheitseinrichtung des Landes verzeichnete von Januar bis Mai 2020 einen Rückgang der Servicenutzung um bis zu 65%. Die Wissenschaftler befürchten, wenn dieser Trend anhalte, könnten die bereits erzielten Erfolge bei der Müttergesundheit zunichte gemacht werden. Kontinuierliche Investitionen in den Ausbau der Gesundheitsinfrastruktur seien nötig, um sie für diese und zukünftige Krisen besser zu wappnen.
Pandemie verstellt den Blick auf andere Krankheiten
Während das Gesundheitssystem damit beschäftigt ist, die Ausbreitung von Covid-19 zu bekämpfen, würden andere lokale Krankheitsausbrüche sträflich vernachlässigt, beklagen Derrick Mensah und KollegInnen.[7] Der Ghana Health Service habe am 15. April 2020 409 Fälle von zerebrospinaler Meningitis gemeldet. Fünf der 16 Provinzen Ghanas waren von der Epidemie betroffen. 40 Menschen seien allein in der zweiten Aprilwoche gestorben – bei weitem mehr also als die 9 Corona-Toten im gleichen Zeitraum. Der Ausbruch sei nicht nur besonders schwerwiegend gewesen. Besorgniserregend sei auch die fehlende Reaktion der Behörden, die der derzeitigen Überlastung des Gesundheitswesens geschuldet sei.
Ursache des Meningitis-Ausbruchs sei ein neuer und gefährlicher bakterieller Erregertypus. 10-15% aller PatientInnen sterben daran, wenn sie nicht behandelt werden. In Ghana waren es wegen fehlender Interventionen viermal so viele Tote. Notwendig sei es, das Gesundheitssystem nachhaltig zu stärken, um eine kontinuierliche Versorgung von PatientInnen auch in Krisenzeiten zu gewährleisten, so die WissenschaftlerInnen.
Massenimpfungen verschoben
Etliche Schlüsselbereiche der Versorgung gerieten durch die Pandemie ins Stocken. Die für April/Mai 2020 geplante Impfkampagne gegen Polio wurde wegen des Verbots von Massenveranstaltungen auf den Herbst verschoben, ebenso die Impfkampagne gegen Gelbfieber und auch die Verteilung von Bettnetzen zum Schutz vor Malaria wurde vorerst ausgesetzt. „Inzwischen sind wir zurück am Start“, so Anthony Adofo Ofosu, Generaldirektor des Ghana Health Service. Man habe gemeinsam mit der WHO Pläne erarbeitet, um essenzielle Gesundheitsdienste auch in Zeiten der Pandemie aufrechtzuerhalten.[8] Dazu zählten Prävention und Behandlung von Infektionskrankheiten, Impfprogramme oder auch die Versorgung vulnerabler Bevölkerungsgruppen wie Kindern und älterer Menschen. Viele Anstrengungen seien unternommen worden, um das Risiko einer Covid-19-Ansteckung im Gesundheitssektor zu minimieren – durch Schulungsprogramme für das Personal, Schutzausrüstung und andere Vorsichtsmaßnahmen. „Wir haben in dieser Zeit viel darüber gelernt wie wir diese Dienstleistungen aufrechterhalten und zugleich sowohl unser Gesundheitspersonal als auch die Öffentlichkeit vor einer Covid-19-Infektion schützen können.“
Artikel aus dem Pharma-Brief 7/2021, S.5
Bild © Owula kpakpo
[1]WHO (2021) Coronavirus (COVID-19) Dashboard https://covid19.who.int/region/afro/country/gh [Zugriff 31.8.2021]
[2]Covax beliefert erstes Land mit Impfstoff. Tagesschau, 24.2.21 www.tagesschau.de/ausland/afrika/ghana-corona-impfstoff-covax-101.html [Zugriff 28.7.21]
[3]Afulani PA et al. (2021) Inadequate preparedness for response to COVID-19 is associated with stress and burnout among healthcare workers in Ghana. PLoS ONE; 16 p e0250294. https://doi.org/10.1371/journal.pone.0250294
[4]www.afro.who.int/countries/ghana [Zugriff 28.7.21]
[5]www.who.int/data/gho/data/indicators/indicator-details/GHO/maternal-mortality-ratio-(per-100-000-live-births) [Zugriff 28.7.21]
[6]Agbozo F and Jahn A (2021) COVID-19 in Ghana: challenges and countermeasures for maternal health service delivery in public health facilities. Reprod. Health;18, p 151 https://doi.org/10.1186/s12978-021-01198-5
[7] Mensah D et al. (2020) COVID-19 effects on national health system response to a local epidemic: the case of cerebrospinal meningitis outbreak in Ghana. Pan African Medical Journal. 35(2):14. https://doi.org/10.11604/pamj.supp.2020.35.2.23138
[8]WHO regional office for Africa (2020) Reinforcing key health services amid COVID-19. www.afro.who.int/news/reinforcing-key-health-services-amid-covid-19 [Zugriff 28.7.21]
Covid-19 – Patente kein Hindernis?
Globaler Zugang zu Impfungen und geistige Eigentumsrechte
 Covid-19-Impfstoffe sind derzeit ein knappes Gut. Die Patentinhaber entscheiden alleine, wo und wieviel Impfstoffe hergestellt werden und wer sie bekommt. Eine Ausweitung der Produktion durch Generikahersteller wäre naheliegend. Doch die Industrie lehnt das in der Welthandelsorganisation diskutierte Aussetzen von geistigen Eigentumsrechten ebenso ab wie freiwillige Lizenzen und Technologietransfer durch den WHO-Patentpool C-TAP.[1] Patente seien nicht das Problem, behauptet Big Pharma.
Covid-19-Impfstoffe sind derzeit ein knappes Gut. Die Patentinhaber entscheiden alleine, wo und wieviel Impfstoffe hergestellt werden und wer sie bekommt. Eine Ausweitung der Produktion durch Generikahersteller wäre naheliegend. Doch die Industrie lehnt das in der Welthandelsorganisation diskutierte Aussetzen von geistigen Eigentumsrechten ebenso ab wie freiwillige Lizenzen und Technologietransfer durch den WHO-Patentpool C-TAP.[1] Patente seien nicht das Problem, behauptet Big Pharma.
Es ist keine Frage, dass es bei Covid-19 global ungerecht zugeht. Auf zehn Länder entfallen 75% der bislang verimpften Dosen, während in 130 Ländern bis Ende Februar noch niemand geimpft wurde. Nicht nur eine gerechtere Verteilung, vor allem auch mehr Impfdosen wären notwendig.
Wie das am besten erreicht werden kann, ist derzeit ein heiß diskutiertes Thema. Bei einer Anhörung im Gesundheitsausschuss des Deutschen Bundestages prallten am 24.2.2021 die Meinungen aufeinander.[2] Dr. Siegfried Throm, der als Vertreter der großen Pharmafirmen für den Verband forschender Arzneimittelunternehmen (VfA) sprach, versuchte in Verdrehung der Tatsachen das Problem kleinzureden: „Wo ist der Bedarf? Es dauert ein bisschen. Aber das gilt nicht nur für die Länder in Europa oder auch in anderen Teilen, sondern das gilt natürlich weltweit.“
VertreterInnen der Pharma-Kampagne, Ärzte ohne Grenzen und Medico International, die bei der Anhörung ebenfalls zugegen waren, betonten dagegen, dass es versäumt wurde, rechtzeitig Voraussetzungen für die globale Massenproduktion von Impfstoffen und Medikamenten zu schaffen. Es sei jetzt dringend notwendig, Patente freizugeben und Technologietransfer zu leisten, um weiteren Herstellern die Produktion von Impfstoffen zu ermöglichen.
Bayer – Zweierlei Maß bei Essure®
Betroffene brasilianische Frauen gehen leer aus
Vor 20 Jahren kam das Verhütungsmittel Essure auf den Markt. Die Spiralen, die in die Eileiter eingeführt wurden, sollten als Ersatz einer chirurgischen Sterilisation dienen und angeblich risikoärmer sein. Doch die Erwartungen erfüllten sich nicht, viele Frauen klagten über Schmerzen oder starke Blutungen, teilweise brachen die Spiralen und mussten operativ entfernt werden. 2018 stellte Bayer den Vertrieb von Essure in Europa[1] und ein Jahr später auch in den USA ein. Zu diesem Zeitpunkt waren in den USA schon Zehntausende Klagen von geschädigten Frauen gegen den Hersteller anhängig. Im August 2020 zahlte Bayer den Betroffenen in den USA 1,35 Milliarden Euro – ohne aber seine Schuld anzuerkennen.[2]
Ganz anders in Brasilien, wo ebenfalls etliche Frauen erhebliche Nebenwirkungen durch Essure erlitten hatten. Hier lehnt Bayer Entschädigungen ab, wie die Tagesschau auf Nachfrage erfuhr. Die Firma begründet ihr unterschiedliches Verhalten damit, dass „die Einigung in den USA eine kommerzielle Entscheidung widerspiegelt, die zum großen Teil von den einzigartigen Aspekten des US-amerikanischen Rechtssystem abhängt, einschließlich der hohen Kosten von US-Rechtsstreitigkeiten.“ [3] Man kann das auch so lesen: Wo es eine patientinnenfreundliche Gesetzgebung gibt, wird bei Schäden gezahlt, wo nicht, zeigt man den Betroffenen die kalte Schulter. 300 brasilianische Frauen wollen jetzt den Konzern in Deutschland verklagen. (JS)
Der Artikel erschien zuerst in GPSP 6/2021
Artikel aus dem Pharma-Brief 8-9/2021, S. 6
[1] In Deutschland ist die Sterilisation eine seltene Verhütungsmethode, und vermutlich wurden hierzulande nur einigen hundert Frauen Essure-Spiralen eingesetzt.
[2] FAZ (2020) Bayer zahlt Klägerinnen in Amerika 1,35 Milliarden Euro. 20.8. www.faz.net/aktuell/wirtschaft/unternehmen/essure-bayer-zahlt-klaegerinnen-in-usa-1-35-milliarden-16914554.html [Zugriff 7.10.2021]
[3] Ebert M (2021) Wir waren Versuchskaninchen. Tageschau.de 25.9. www.tagesschau.de/wirtschaft/unternehmen/bayer-essure-klagen-101.html [Zugriff 7.10.2021]
40 Jahre BUKO Pharma-Kampagne
Mehr als ein Grund zu feiern
Am Ende unserer ABR-Konferenz gab es eine kleine virtuelle Feier zum vierzigjährigen Bestehen der Pharma-Kampagne. Eingeleitet wurde die Veranstaltung mit einem kurzen Film über Highlights der Kampagnenarbeit aus den letzten 40 Jahren, den Sie auf unserer Website anschauen können. Produziert hat ihn unser ehemaliger Mitarbeiter Stefan Jankowiak.
Dann folgte – moderiert von unserer Vorstandsfrau Elisabeth Lipsewers – ein bunter Regen von Glückwünschen aus aller Welt. Gopal Dabade vom Drug Action Forum Karnataka, Indien, hob hervor, wie wichtig die Arbeit von BUKO angesichts des massiven negativen Einflusses großer Pharmakonzerne im globalen Süden sei. Andy Gray aus Südafrika lobte die Zusammenarbeit beim Kampf für den Zugang zu Aids-Medikamenten. Das unterstrich auch Peter Wiesner vom deutschen Aktionsbündnis gegen Aids.
Davina Dietrich und Jan Quakernack von unserem Straßentheater „Schluck & weg“ lieferten eine geschickt inszenierte Botschaft. Sigrun Landes-Brenner grüßte von Brot für die Welt, Alexander Lohner von Misereor. Wilbert Bannenberg erinnerte humorvoll an die nachbarschaftliche Entwicklungshilfe: Der niederländische Pragmatismus hat sicher zum Überleben der Kampagne erheblich beigetragen.
Der entfernteste Gruß kam von Anwar Fazal aus Malaysia, der an die gemeinsame Gründung des Netzwerks Health Action International (HAI) vor 40 Jahren in Genf erinnerte. Birte Bogatz-Mander vom globalen HAI-Büro in Amsterdam hob die Hartnäckigkeit und Ausdauer der Pharma-Kampagne hervor und wünschte viele weitere Jahre guter Zusammenarbeit.
Dies ist nur eine kleine Auswahl der vielen Highlights des Abends. Zahlreiche Gratulationen erreichten uns auch auf schriftlichem Weg. Ihnen und euch allen ganz herzlichen Dank dafür!
Und fest versprochen sei schon hier, dass es eine echte Feier geben wird, wenn das wieder möglich ist. Wie bei der Pharma-Kampagne üblich, werden wir das Fest mit einem thematischen Input verbinden – aber einfach nur zum Feiern zu kommen, ist auch völlig OK. (JS)
Bild © Jörg Schaaber
Artikel aus dem Pharma-Brief 5/2021, S.7